High Efficient of the Intermolecular Radical Reactions through three-Component Carbo-Oximation Process using new ready available Sulfonyl oxime.
Shireen Mohammed, Maher Khalid
Department of Chemistry, Collage of Science, Zakho University, Kurdistan – Region, Iraq.
DOI : http://dx.doi.org/10.13005/ojc/310308
Article Received on :
Article Accepted on :
Article Published : 10 Aug 2015
Addition of functionalized carbon fragments across the olefinic π-system through a free-radical carbo-oximation process offers a straightforward access to valuable intermediates for organic synthesis. For this purpose, we designed a new protected sulfonyl oxime, which enable rapid radical addition with high yields under mild conditions.
KEYWORDS:Carbo-oximation processes; free three multicomponent reactions; sulfonyl oxime acceptor
Download this article as:| Copy the following to cite this article: Mohammed S, Khalid M. High Efficient of the Intermolecular Radical Reactions through three-Component Carbo-Oximation Process using new ready available Sulfonyl oxime. Orient J Chem 2015;31(3). |
| Copy the following to cite this URL: Mohammed S, Khalid M. High Efficient of the Intermolecular Radical Reactions through three-Component Carbo-Oximation Process using new ready available Sulfonyl oxime. Orient J Chem 2015;31(3). Available from: http://www.orientjchem.org/?p=10259 |
Introduction
Free radical multi component reactions (MCR) are more flexible and fulfill for an efficient carbo-functionalization of olefins1. Radical MCRs have thus attracted a considerable attention. More recently, the addition of carbon fragments across the π-bond of non-activated olefins through free-radical pathways has received intense scrutiny, resulting in the description of useful transformations, such as for instance carbo-alkenylation2,3, carbo-alkynylation4 and carbo-allylation of olefins. These processes also illustrate the importance of the influence of polar effects in free-radical MCRs5. Various electrophilic species may be envisioned, but sulfones including vinyl6-,alkynyl-, allyl7– and azido-sulfones8 hold a prominent place, due to the fast and efficient β-fragmentation of the sulfonyl moiety. Sulfones also allow to install other R3 substituents on the olefinic backbone such as (SPh, CN, N3, Cl, etc..)9. Kim and co-workers10 have thus shown that sulfonyl oximes are also excellent radical acceptors11, enabling the incorporation of an oxime onto a carbon framework as electrophilic radical traps. The strategy of the present work relies on the generalization of carbo-oximation of olefins as a formal carbo-formylation process. Carbo-formylation of an olefin under radical conditions has been described by Ryu et al. using carbon monoxide as the radical trap12. The use of Kim’s sulfonyl oxime constitutes a more practical surrogate to toxic CO13, which generally requires relatively high pressure, and therefore specific autoclave equipment14.
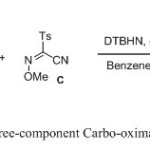 |
Scheme 1 Click here to View scheme |
Experimental
General: Equipment, Chemicals and Work Technique
All reactions were carried out under argon atmosphere with dry solvents under anhydrous conditions. Yields refer to chromatographically and spectroscopically (1H NMR) homogeneous materials. Commercial reagents were used without purification. Benzene was distilled over sodium and benzophenone. DCE were distilled from CaH2.1H NMR and 13C NMR were recorded on Bruker DPX-200 FT (1H: 200 MHz, 13C: 50.3 MHz), Bruker Advance- 300FT (1H: 300 MHz, 13C: 75.5 MHz),. All NMR spectra present in this work were measured in CDCl3 solution. All chemical shifts are given in ppm. The chemical shifts (δ) and coupling constants (J) are expressed in ppm and Hz respectively. The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, b = broad. High resolution mass spectra were recorded on a Micromass ZABSpec TOF, on a Q-Tof Applied Biosystems and on Waters Q-Tof 2 apparatus. IR spectra were recorded on a Perkin-Elmer 1710 spectrophotometer or on a Perkin-Elmer aragon 1000 FT-IR spectrophotometer. Thin Layer Chromatography (TLC): Merck Kieselgel 60 F254 on aluminium foil from Macherey-Nagel. Detection was carried out under UV light at 254 nm and 365 nm. Column chromatography was performed with Merck Silica Gel 60 (70-230 mesh), (230-400 mesh ASTM) and Baker silica gel (0.063-0.200 mm) were used for flash chromatography.
General procedure for the three-component carbo-oximation with sulfonyl oxime (C) (Table 1). Three-component adducts (11-20).
In a dry two-neck round-bottom flask equipped with a condenser and a magnetic stirrer were successively added oxime C (2 equiv, See Table 1), iodoester A (1 equiv) and the desired alkene partner B (4 equiv) in benzene (0.4 M). Argon was bubbled directly into the flask for 30 min. (Bu3Sn)2 (1.5 equiv) was injected and the flask was heated to 60oC. DTBHN was added after 5 min, then every 90 min if required (TLC). After total consumption of the starting iodide, the resulting mixture was concentrated in vacuo and purified by silica gel chromatography (Petroleum ether/ EtOAc) to afford the desired product.
tert-butyl-2-(cyano(methoxyimino)methyl)-3-(2-ethoxy-2-oxoethyl)piperidine-1-carboxylate (11)
Compound 11 was obtained according to the general procedure described above from ethyl iodoacetate A (53 mg, 0.25 mmol, 1 equiv), sulfonyl oxime C (119 mg, 0.5 mmol, 2 equiv), tert-butyl 3,4-dihydropyridine-1(2H)-carboxylate 1 (197 mg, 1 mmol, 4 equiv), (Bu3Sn)2 (0.19 mL,0.38 mmol), and DTBHN (4 mg, 0.04 mmol, 10 mol %) added by 5 mol % portion every 1.5 h, in degassed benzene (1.5 mL). Concentration in vacuo, followed by purification by flash chromatography (silica gel, 90/10 PE / EtOAc) afforded 11 (69 mg, 78%) as a colorless oil. Rf= 0.4 (PE/EtOAc 90/10). IR (ATR) νmax (cm-1) = 2957, 2855, 2711, 1732, 1690, 1513, 1278, 939.1H NMR (CDCl3, 300 MHz): δ (ppm) = 4.91 (d, 1H, J = 6 Hz, CH), 4.18 (q, 2H J = 3 and 6 Hz, CH2), 4.02 (s, 3H, CH3), 3.94-3.89 (m, 1H, CH), 3.39-3.34 (m, 1H, CH), 2.43-2.34 (m, 2H, CH2), 2.18-2.11 (m, 2H, CH2), 1.91-1.77 (m, 2H, CH2), 1.45 (s, 9H, 3CH3), 1.38 (appearant t, 3H, J = 6 and 3 Hz, CH3), 1.25-1.19 (m, 1H, CH).13C NMR (CDCl3, 75.5 MHz): δ (ppm) = 174.9 (C=O ester), 156.9 (C=O ester), 138.4 (C), 105.5 (CN), 81.0 (C),61.2 (C), 60.6 (C), 60.4 (C), 43.3 (C), 37.3 (C), 36.3 (C), 28.4 (C), 26.8 (C), 23.3 (C), 14.7 (C). HRMS (ESI): [M+H]+ C17H28N3O5 calcd. 354.1993, found 354.19330.
ethyl 2-(2-(cyano(methoxyimino)methyl)tetrahydro-2H-pyran-3-yl)acetate (12)
Compound 12 was obtained according to the general procedure described above from ethyl iodoacetate A (53 mg, 0.25 mmol, 1 equiv), sulfonyl oxime C (119 mg, 0.5 mmol, 2 equiv), 3,4-dihydro-2H-pyran 2 (84 mg, 1 mmol, 4 equiv), (Bu3Sn)2 (0.19 mL,0.38 mmol), and DTBHN (4 mg, 0.04 mmol, 10 mol %) added by 5 mol % portion every 1.5 h, in degassed benzene (1.5 mL). Concentration in vacuo, followed by purification by flash chromatography (silica gel, 88/12 PE / EtOAc) afforded 12 (64 mg, 81%) as a colorless oil. Rf= 0.4 (PE/EtOAc 88/12). IR (ATR) νmax (cm-1) = 2970, 1730, 1652, 1447, 1308, 1149, 1085, 688..1H NMR (CDCl3, 300 MHz): δ (ppm) = 5.17 (d, 1H, J = 6 Hz, CH), 4.17 (appearant q, 2H J = 3 and 6 Hz, CH2), 3.99 (s, 3H, CH3), 3.80-3.76 (m, 1H, CH), 3.71-3.67 (m, 1H, CH), 2.91-2.82 (m, 1H, CH), 2.30-2.26 (m, 1H, CH), 2.23-2.16 (m, 1H, CH), 2.06-2.01(m, 1H, CH), 1.38 (appearant t, 3H, J = 6 and 3 Hz, CH3), 1.20-1.13 (m, 1H, CH).13C NMR (CDCl3, 75.5 MHz): δ (ppm) = 174.9 (C=O ester), 132.1 (C), 110.8 (CN), 75.5 (C), 67.5 (C),61.2 (C), 60.6 (C), 60.4 (C), 36.8 (C), 34.8 (C), 25.9 (C), 24.7 (C), 14.7 (C). HRMS (ESI): [M+H]+ C12H19N2O4 calcd. 255.1345, found 252.1344.
ethyl 4-(cyano(methoxyimino)methyl)decanoate (13)
Compound 13 was obtained according to the general procedure described above from ethyl iodoacetate A (53 mg, 0.25 mmol, 1 equiv), sulfonyl oxime C (119 mg, 0.5 mmol, 2 equiv), oct-1-ene 3 (112 mg, 1 mmol, 4 equiv), (Bu3Sn)2 (0.19 mL,0.38 mmol), and DTBHN (4 mg, 0.04 mmol, 10 mol %) added by 5 mol % portion every 1.5 h, in degassed benzene (1.5 mL). Concentration in vacuo, followed by purification by flash chromatography (silica gel, 95/5 PE / EtOAc) afforded 13 (64 mg, 91%) as a colorless oil. Rf= 0.4 (PE/EtOAc 95/5). IR (ATR) νmax (cm-1) = 2976, 2957, 2855, 2711, 1732, 1690, 1513, 1278, 939.1H NMR (CDCl3, 300 MHz): δ (ppm) = 4.17 (q, 2H, J = 3 Hz, CH2), 4.06-4.03 (m, 1H CH), 4.00 (s, 3H, CH3), 2.30 (appearant t, 2H, J = 3 and 6 Hz, CH2), 2.00-1.96 (m, 1H, CH), 1.92-1.88 (m, 1H, CH), 1.57-1.55 (m, 2H, CH2), 1.48-1.31 (m, 1H, CH and 5CH2), 0.99 (appearant t, 3H, J = 6 and 3 Hz, CH3).13C NMR (CDCl3, 75.5 MHz): δ (ppm) = 174.3 (C=O ester), 135.3 (C), 114.9 (CN), 61.2 (C), 60.4 (C),34.2 (C), 32.9 (C), 32.6 (C), 31.6 (C), 29.9 (C), 29.3 (C), 27.8 (C), 22.9 (C), 14.7 (C), 14.0 (C). HRMS (ESI): [M+H]+ C15H27N2O7 calcd. 283.2021, found 283.2021.
ethyl 3-(1-(cyano(methoxyimino)methyl)cyclohexyl)propanoate (14)
Compound 14 was obtained according to the general procedure described above from ethyl iodoacetate A (53 mg, 0.25 mmol, 1 equiv), sulfonyl oxime C (119 mg, 0.5 mmol, 2 equiv), methylenecyclohexane 4 (96 mg, 1 mmol, 4 equiv), (Bu3Sn)2 (0.19 mL,0.38 mmol), and DTBHN (4 mg, 0.04 mmol, 10 mol %) added by 5 mol % portion every 1.5 h, in degassed benzene (1.5 mL). Concentration in vacuo, followed by purification by flash chromatography (silica gel, 94/6 PE / EtOAc) afforded 14 (59 mg, 89%) as a colorless oil. Rf= 0.36 (PE/EtOAc 94/6). IR (ATR) νmax (cm-1) = 1680, 1305, 1145, 1020, 987.1H NMR (CDCl3, 300 MHz): δ (ppm) = 4.17 (q, 2H, J = 3 Hz, CH2), 4.00 (s, 3H CH3), 2.29 (appearant t, 2H, J = 3 and 6 Hz, CH2), 1.91 (appearant t, 2H, J = 3 and 6 Hz, CH2), 1.87-1.82 (m, 2H, CH2), 1.73-1.53 (m, 8H, 4CH2), 1.37 (m, 2H, CH2), 1.73-1.53 appearant t, 3H, J = 6 and 3 Hz, CH3).13C NMR (CDCl3, 75.5 MHz): δ (ppm) = 174.4 (C=O ester), 127.7 (C), 116.2 (CN), 61.2 (C), 60.4 (C),49.5 (C), 33.0 (C), 30.6 (C), 28.1 (C), 25.6 (C), 22.2 (C), 14.7 (C). HRMS (ESI): [M+H]+ C14H23N2O3 calcd. 267.1708, found 267.1709.
ethyl 3-(1-(cyano(methoxyimino)methyl)cyclopentyl)propanoate (15)
Compound 15 was obtained according to the general procedure described above from ethyl iodoacetate A (53 mg, 0.25 mmol, 1 equiv), sulfonyl oxime C (119 mg, 0.5 mmol, 2 equiv), methylenecyclopentane 5 (68 mg, 1 mmol, 4 equiv), (Bu3Sn)2 (0.19 mL,0.38 mmol), and DTBHN (4 mg, 0.04 mmol, 10 mol %) added by 5 mol % portion every 1.5 h, in degassed benzene (1.5 mL). Concentration in vacuo, followed by purification by flash chromatography (silica gel, 94/6 PE / EtOAc) afforded 15 (55 mg, 87%) as a colorless oil. Rf= 0.36 (PE/EtOAc 94/6). IR (ATR) νmax (cm-1) = 1690, 1304, 1140, 1015, 980.1H NMR (CDCl3, 300 MHz): δ (ppm) = 4.17 (appearant q, 2H, J = 3 and 6 Hz, CH2), 3.99 (s, 3H CH3), 2.36 (appearant t, 2H, J = 3 and 6 Hz, CH2), 2.34 (appearant t, 2H, J = 3 and 6 Hz, CH2), 1.91-1.80 (m, 2H, CH2), 1.72-1.54 (m, 6H, 3CH2), 1.38 (t, 3H, J = 3 Hz, CH3).13C NMR (CDCl3, 75.5 MHz): δ (ppm) = 174.4 (C=O ester), 127.6 (C), 116.5 (CN), 61.2 (C), 60.4 (C),59.9 (C), 36.1 (C), 30.6 (C), 30.2 (C), 23.6 (C), 14.7 (C). HRMS (ESI): [M+H]+ C13H21N2O3 calcd. 253.1552, found 253.1553.
ethyl 4-(cyano(methoxyimino)methyl)-4-ethylhexanoate (16)
Compound 16 was obtained according to the general procedure described above from ethyl iodoacetate A (53 mg, 0.25 mmol, 1 equiv), sulfonyl oxime C (119 mg, 0.5 mmol, 2 equiv), 3-methylenepentane 6 (84 mg, 1 mmol, 4 equiv), (Bu3Sn)2 (0.19 mL,0.38 mmol), and DTBHN (4 mg, 0.04 mmol, 10 mol %) added by 5 mol % portion every 1.5 h, in degassed benzene (1.5 mL). Concentration in vacuo, followed by purification by flash chromatography (silica gel, 94/6 PE / EtOAc) afforded 16 (59 mg, 92%) as a colorless oil. Rf= 0.35 (PE/EtOAc 94/6). IR (ATR) νmax (cm-1) = 1679, 1300, 1148, 1018, 980.1H NMR (CDCl3, 300 MHz): δ (ppm) = 4.43 (appearant t, 2H, J = 3 and 6 Hz, CH2), 4.22 (q, 2H, J = 3 Hz, CH2), 4.02 (s, 3H, CH3), 2.06 (appearant t, 2H, J = 3 and 6 Hz, CH2), 1.84 (appearant q, 2H, 2H, J = 3 and 6 Hz, CH2), 1.58 (appearant q, 2H, J = 3 and 6 Hz, CH2), 1.39 (t, 3H, J = 3 Hz, CH3), 1.01 (t, 6H, J = 3 Hz, CH3).13C NMR (CDCl3, 75.5 MHz): δ (ppm) = 174.4 (C=O ester), 126.4 (C), 116.3 (CN), 61.2 (C), 60.4 (C),51.0 (C), 30.6 (C), 27.5 (C), 27.4 (C), 14.7 (C), 8.2 (C). HRMS (ESI): [M+H]+ C13H23N2O3 calcd. 255.1708, found 255.17080.
ethyl 5-cyano-5-(methoxyimino)-4-((trimethylsilyl)methyl)pentanoate (17)
Compound 17 was obtained according to the general procedure described above from ethyl iodoacetate A (53 mg, 0.25 mmol, 1 equiv), sulfonyl oxime C (119 mg, 0.5 mmol, 2 equiv), allyltrimethylsilane 7 (11.4 mg, 1 mmol, 4 equiv), (Bu3Sn)2 (0.19 mL,0.38 mmol), and DTBHN (4 mg, 0.04 mmol, 10 mol %) added by 5 mol % portion every 1.5 h, in degassed benzene (1.5 mL). Concentration in vacuo, followed by purification by flash chromatography (silica gel, 96/4 PE / EtOAc) afforded 17 (68 mg, 95%) as a colorless oil. Rf= 0.35 (PE/EtOAc 96/4). IR (ATR) νmax (cm-1) = 1700, 1670, 1150, 1201, 1015, 940.1H NMR (CDCl3, 300 MHz): δ (ppm) = 4.14 (q, 2H, J = 3 Hz, CH2), 4.02 (s, 3H, CH3), 3.53-3.48 (m, 1H, CH), 2.49 (appearant t, 2H, J = 3 and 6 Hz, CH2), 1.92-1.84 (m, 2H, 2H, CH2), 1.37 (appearant t, 3H, J = 3 and 6 Hz, CH3), 1.03-0.99 (m, 1H, CH), 0.74 (m, 1H, CH), 0.27 (s, 9H, 3CH3).13C NMR (CDCl3, 75.5 MHz): δ (ppm) = 174.3 (C=O ester), 130.1 (C), 114.9 (CN), 61.2 (C), 60.4 (C),37.2 (C), 32.6 (C), 30.2 (C), 14.7 (C), 14.1 (C). HRMS (ESI): [M+H]+ C13H25N2O3Si calcd. 285.1634, found 285.1634.
ethyl 5-cyano-4-ethoxy-5-(methoxyimino)pentanoate (18)
Compound 18 was obtained according to the general procedure described above from ethyl iodoacetate A (53 mg, 0.25 mmol, 1 equiv), sulfonyl oxime C (119 mg, 0.5 mmol, 2 equiv), ethoxyethene 8 (72 mg, 1 mmol, 4 equiv), (Bu3Sn)2 (0.19 mL,0.38 mmol), and DTBHN (4 mg, 0.04 mmol, 10 mol %) added by 5 mol % portion every 1.5 h, in degassed benzene (1.5 mL). Concentration in vacuo, followed by purification by flash chromatography (silica gel, 90/10 PE / EtOAc) afforded 18 (51 mg, 85%) as a colorless oil. Rf= 0.35 (PE/EtOAc 90/10). IR (ATR) νmax (cm-1) = 1700, 1680, 1630, 1307, 1144, 1070.1H NMR (CDCl3, 300 MHz): δ (ppm) = 4.85 (appearant t, 1H, J = 3 and 6 Hz, CH), 4.17 (q, 2H, J = 3 Hz, CH2), 2.38 (appearant t, 2H, J = 3 and 6 Hz, CH2), 2.19-2.09 (m, 2H, CH2), 1.38 (t, 3H, J = 3 Hz, CH3), 1.20 (appearant t, 3H, J = 3 and 6 Hz, CH3).13C NMR (CDCl3, 75.5 MHz): δ (ppm) = 174.3 (C=O ester), 135.4 (C), 109.5 (CN), 74.0 (C), 64.6 (C),61.2 (C), 60.4 (C), 31.0 (C), 30.6 (C), 15.5 (C), 14.7 (C). HRMS (ESI): [M+H]+ C11H19N2O4 calcd. 243.1345, found 243.1345.
3-(cyano(methoxyimino)methyl)-6-ethoxy-3-methyl-6-oxohexyl benzoate (19)
Compound 19 was obtained according to the general procedure described above from ethyl iodoacetate A (53 mg, 0.25 mmol, 1 equiv), sulfonyl oxime C (119 mg, 0.5 mmol, 2 equiv), 3-methylbut-3-en-1-yl benzoate 9 (72 mg, 1 mmol, 4 equiv), (Bu3Sn)2 (0.19 mL,0.38 mmol), and DTBHN (4 mg, 0.04 mmol, 10 mol %) added by 5 mol % portion every 1.5 h, in degassed benzene (1.5 mL). Concentration in vacuo, followed by purification by flash chromatography (silica gel, 92/8 PE / EtOAc) afforded 19 (71 mg, 79%) as a colorless oil. Rf= 0.37 (PE/EtOAc 92/8). IR (ATR) νmax (cm-1) = 2955, 1750, 1733, 1446, 1430, 1300, 1280, 1080, 750.1H NMR (CDCl3, 300 MHz): δ (ppm) = 7.94 (d, 2H, CHar), 7.48-7.37 (m, 3H, CHar), 4.24 (appearant t, 2H, J = 3 and 6 Hz, CH2), 4.16 (q, 2H, J = 3 Hz, CH2), 3.99 (s, 3H, CH3), 2.31 (t, 2H, J = 6 Hz, CH2), 2.13-2.04 (m, 4H, 2CH2), 1.32 (s, 3H, CH3).13C NMR (CDCl3, 75.5 MHz): δ (ppm) = 174.4 (C=O ester), 167.0 (C=O ester), 133.0 (C), 130.2 (Car), 129.6 (Car), 128.8 (Car), 113.2 (CN), 64.4 (C), 61.2 (C),60.4 (C), 38.7 (C), 36.8 (C), 30.5 (C), 29.7 (C), 25.0 (C), 14.7 (C). HRMS (ESI): [M+H]+ C19H25N2O5 calcd. 361.1763, found 361.17630.
ethyl 4-(cyano(methoxyimino)methyl)-6-hydroxy-4-methylhexanoate (20)
Compound 20 was obtained according to the general procedure described above from ethyl iodoacetate A (53 mg, 0.25 mmol, 1 equiv), sulfonyl oxime C (119 mg, 0.5 mmol, 2 equiv), 3-methylbut-3-en-1-ol 10 (86 mg, 1 mmol, 4 equiv), (Bu3Sn)2 (0.19 mL,0.38 mmol), and DTBHN (4 mg, 0.04 mmol, 10 mol %) added by 5 mol % portion every 1.5 h, in degassed benzene (1.5 mL). Concentration in vacuo, followed by purification by flash chromatography (silica gel, 94/6 PE / EtOAc) afforded 20 (47 mg, 76%) as a colorless oil. Rf= 0.34 (PE/EtOAc 90/10). IR (ATR) νmax (cm-1) = 2920, 1730, 1650, 1315, 1201, 1105, 939.1H NMR (CDCl3, 300 MHz): δ (ppm) = 4.16 (appearant q, 2H, J = 3 and 6 Hz, CH2), 4.01 (s, 3H, CH3), 3.49 (appearant t, 2H, J = 3 and 6 Hz, CH2), 2.31 (appearant t, 2H, J = 3 and 6 Hz, CH2), 2.01 (t, 2H, J = 6 Hz, CH2), 1.85 (appearant t, 2H, J = 3 and 6 Hz, CH2), 1.37 (t, 3H, J = 3 Hz, CH3), 1.31 (s, 3H, CH3), 0.56 (s, 1H, OH).13C NMR (CDCl3, 75.5 MHz): δ (ppm) = 174.4 (C=O ester), 128.8 (C), 113.2 (CN), 61.2 (C), 60.4 (C),59.9 (C), 38.7 (C), 38.0 (C), 30.5 (C), 29.7 (C), 25.0 (C), 14.7 (C). HRMS (ESI): [M+H]+ C12H21N2O4 calcd. 257.1501, found 257.1501.
Results and Disussion
The 3-component carbo-oximation process was first carried out using new sulfonyl oxime, ready available from the corresponding α-sulfonyl nitrile15. Such reactions proceed through the preliminary decomposition of DTBHN initiator to produce nitrogen gas and tBuO radical. Addition of the latter onto tin compound produced tin radical. Tin radical can then abstract the iodide group from ethyliodoacetate (A) and form the electrophilic radical (Ai). Addition of an electron-poor radical to the less hindered end of an olefin (B), forming a new nucleophilic radical (Bi), which can then be trapped by an electrophilic partner such as cyanosulfonyl oxime (C), affording the expected products (P) and expel the tosylate group through β-fragmentation process to propagate the radical chain Scheme 2.
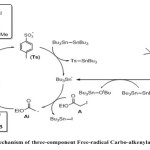 |
Scheme 2 Click here to View scheme |
The 3-component carbo-oximation process was first carried out using cyano sulfonyl oxime B (2 eq.), an excess of the olefins 1-10 (4 eq.), ethyliodoester as precursor A (1 eq.), and (Bu3Sn)2 (1.5 equiv) in benzene (degassed) as a solvent. The reaction was initiated using di-tert-butylhyponitrite (DTBHN) (10 mol %). The results are summarized in Scheme 3 (Table 1) below. Generally good and reproducible yields of the 3-component adducts 11-20 were obtained (76-95%). The final oximes were easily isolated through chromatography over silica gel. A broad variety of substituents on the olefin is compatible with the reaction conditions. The reaction conditions were found to be compatible with Boc-protected amines and dihydro-pyran as an electron-rich olefins (entry 1 and 2). The iodoester substrate A was also shown to react efficiently with normal olefin and methylene cyclohexene as well as methylene cyclopentene (entry 3-6). The reaction was extended with using electron-rich olefins such as allylsilanes and vinylether, which produced the expected products 17 and 18 in very good yields (entry 7 and 8). Generation of products 19 and 20 with quaternary center carbon was also obtained with a good yield (entry 9 and 10).
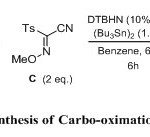 |
Scheme 3 Click here to View scheme |
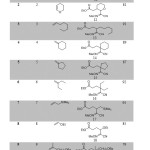 |
Table 1: Three-component Carbo-oximation of various Olefins using sulfonyl oxime nitrile. |
Conclusion
In summary, a sequential carbo-oximation protocol involving a three-component radical process was developed under mild conditions in a single pot starting from readily available ehtyliodoacetate, different olefins and new sulfonyl oxime acceptor. This three-component reaction proceed through the addition of a radical species derived from a iodoester and a vinylsulfone across the olefinic groups backbone and formation of two new carbon-carbon bonds,
which enables facile hydrolysis of the oxime under mild conditions after post-functionalization of the carbo-oximation products and provides a rapid access to lactones, after Mukaiyama aldol or Sakurai allylation reactions or more complex piperidinones using Pictete-Spengler processes.
Acknowledgements
We gratefully acknowledge (Bordeaux 1) University (France), the Kurdistan Government and the HCDP Program for financial support.
References
- (a) I. Ryu, Free-radical-mediated Multicomponent Coupling Reactions In Multicomponent Reactions; J. Zhu, H.E. Bienaym, Eds.; Wiley-VCH: Weinheim, Germany, 2005; Chapter 6, p 169; (b) I. Ryu; N. Sonoda; D. P. Curran, Chem. Rev. 1996, 96, 177. (a) G. Rouquet; F. Robert; R. Mereau; F.. Castet; Y. Landais. Chem.-Eur. J. 2011, 17, 13904; (b) H. Jasch; Y. Landais; M. r. Heinrich, Chem.-Eur. J. 2013, 19, 841; (c) V. Liautard; F. Robert; Y. Landais, Org. Lett. 2011, 13, 2658; (d) C. Poittevin; V. Liautard; R. Beniaz; F. Robert; Y. Landais, Org. Lett. 2013, 15, 2814.
- For reviews on allylation, vinylation, cyanation, oximation using sulfonyl derivatives, see: (a) Kim, S.; Kim, S. Bull. Chem. Soc. Jpn. 2007, 80, 809; (b) Bertrand, F.; Le Guyader, F.; Liguori, L.; Ouvry, G.; Quiclet-Sire, B.; Seguin, S.; Zard, S. Z. C. R. Acad. Sci. 2001, 4, 547.
- (a) Gong, J.; Fuchs, P. L. J. Am. Chem. Soc. 1996, 118, 4486; (b) Xiang, J.; Fuchs, P. L. J. Am. Chem. Soc. 1996, 118, 11986; (c) Xiang, J.; Jiang, W.; Gong, J.; Fuchs, P. L. J. Am. Chem. Soc. 1997, 119, 4123; (d) Xiang, J.; Fuchs, P. L. Tetrahedron Lett. 1996, 37, 5269; (e) Xiang, J.; Jiang, W.; Fuchs, P. L. Tetrahedron Lett. 1997, 38, 6635; (f) Xiang, J.; Fuchs, P. L. Tetrahedron Lett. 1998, 39, 8597; (g) Uenoyama, Y.; Fukuyama, T.; Morimoto, K.; Nobuta, O.; Nagai, H.; Ryu, I. Helv. Chim. Acta 2006, 89, 2483.
- (a) Ryu, I.; Yamazaki, H.; Ogawa, A.; Kambe, N.; Sonoda, N. J. Am. Chem. Soc. 1993, 115, 1187; (b) Ryu, I.; Muraoka, H.; Kambe, N.; Komatsu, M.; Sonoda, N. J. Org. Chem. 1996, 61, 6396; (c) Schaffner, A. P.; Sarkunam, K.; Renaud, P. Helv. Chim. Acta 2006, 89, 2450.
- Godineau, E.; Landais, Y. Chem.-Eur. J. 2009, 15, 3044.
- (a) Russell, G. A.; Tahtoush, H.; Ngoviwatchai, P. J. Am. Chem. Soc. 1984, 106, 4622; (b) Russell, G. A.; Ngoviwatchai, P. J. Org. Chem. 1989, 54, 1836; (c) Russell, G. A.; Ngoviwatchai, P.; Tashtoush, H. L.; Pla-Dalmau, A.; Khanna, R. J. J. Am. Chem. Soc. 1988, 110, 3530; (d) Bertrand, F.; Quiclet-Sire, B.; Zard, S. Z. Angew. Chem., Int. Ed. 1999, 38, 1943; (e) Schaffner, A.-P.; Darmency, V.; Renaud, P. Angew. Chem., Int. Ed. 2006, 45, 5847.
- (a) Chatgilialoglu, C.; Ballestri, M.; Vecchi, D.; Curran, D. P. Tetrahedron Lett. 1996, 37, 6383; (b) Kim, S.; Cho, C. H.; Uenoyama, Y.; Ryu, I. Synlett 2005, 3160; (c) Kim, S.; Otsuka, N.; Ryu, I. Angew. Chem., Int. Ed. 2005, 44, 6183; (d) Charrier, N.; Zard, S. Z. Angew. Chem., Int. Ed. 2008, 47, 9443; (e) Le Guyader, F.; Quiclet-Sire, B.; Seguin, S.; Zard, S. Z. J. Am. Chem. Soc. 1997, 119, 7410; (f) Quiclet-Sire, B.; Seguin, S.; Zard, S. Z. Angew. Chem., Int. Ed. 1998, 37, 2864.
- (a) Ollivier, C.; Renaud, P. J. Am. Chem. Soc. 2001, 123, 4717; (b) Renaud, P.; Ollivier, C.; Panchaud, P. Angew Chem. 2002, 114, 3610; (c) Ollivier, C.; Renaud, P. Angew. Chem. Int. Ed. 2002, 41, 3460; (d) Panchaud, P.; Ollivier, C.; Renaud, P.; Zigmantas, S. J. Org. Chem. 2004, 69, 2755; (e) Panchaud, P.; Renaud, P. J. Org. Chem. 2004, 69, 3205; (f) Panchaud, P.; Renaud, P. Chimia 2004, 58, 232; (j) Panchaud, P.; Chabaud, L.; Landais, Y.; Ollivier, C.; Renaud, P.; Zigmantas, S. Chem. Eur. J. 2004, 10, 3606
- (a) Roques, N.; Galvez, M.; Bonnefoy, A.; Larquetoux, L.; Spargnol, M. Chim. Oggi-Chem. Today 2003, 21, 43; (b) Cao, L.; Weidner, K.; Renaud, P. Adv. Synth. Catal. 2011, 353, 3467; (c) Schaffner, A.-P.; Montermini, F.; Pozzi, D.; Darmency, V.; Scanlan, E. M.; Renaud, P. Adv. Synth. Catal. 2008, 350, 1163; (d) Weidner, K.; Giroult, A.; Panchaud, P.; Renaud, P. J. Am. Chem. Soc. 2010, 132, 17511; (e) Mantrand, N.; Renaud, P. Tetrahedron 2008, 64, 11860; (f) Panchaud, P.; Ollivier, C.; Renaud, P.; Zigmantas, S. J. Org. Chem. 2004, 69, 2755; (g) Renaud, P.; Ollivier, C.; Panchaud, P. Angew. Chem., Int. Ed. 2002, 41, 3460.
- (a) Kim, S.; Lee, I. Y.; Yoon, J. Y.; Oh, D. H. J. Am. Chem. Soc. 1996, 118, 5138; (b) Kim, S.; Yoon, J.-Y. J. Am. Chem. Soc. 1997, 119, 5982; (c) Kim, S.; Song, H.-J.; Choi, T.-L.; Yoon, J. Y. Angew. Chem., Int. Ed. 2001, 40, 2524; (d) Kim, S.; Yoon, J. Y.; Lee, I. Y. Synlett 1997, 475; (e) Ryu, I.; Kuriyama, H.; Minakata, S.; Komatsu, M.; Yoon, J.-Y.; Kim, S. J. Am. Chem. Soc. 1999, 121, 12190.
- (a) Godineau, E.; Landais, Y. J. Am. Chem. Soc. 2007, 129, 12662; (b) Godineau, E.; Sch€afer, C.; Landais, Y. Org. Lett. 2006, 8, 4871; (c) Godineau, E.; Landais, Y. J. Org. Chem. 2008, 73, 6983.
- Ryu, I.; Muraoka, H.; Kambe, N.; Komatsu, M.; Sonoda, N. J. Org. Chem. 1996, 61, 6936.
- Gaspar, B.; Carreira, E. M. J. Am. Chem. Soc. 2009, 131, 13214.
- Kuniyasu, H.; Ogawa, A.; Higaki, K.; Sonoda, N. Organometallics 1992, 11, 3937.
- (a) van Schoor, A.; Schumann, W.; Lust, S.; Flemming, H. Patent. 1141487, 19600423, 1962. (b) Bayer, H.; Sauter, H.; Benoit, R.; Mueller, R. Ammermann, E.; Lorenz, G. Patent. 94-118383 656352, 19941123, 1995.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


