DFT study on the covalent adsorption of drug carvedilol onto COOH functionalized carbon nanotubes
M. Rahbar, A. Morsali *, M. R. Bozorgmehr and S. A. Beyramabadi
Department of Chemistry, Mashhad Branch, Islamic Azad University, Mashhad, Iran & Research Center for Animal Development Applied Biology, Mashhad Branch, Islamic Azad University, Mashhad 917568, Iran. Correspondence Author E-mail: almorsali@yahoo.com
DOI : http://dx.doi.org/10.13005/ojc/310318
Article Received on :
Article Accepted on :
Article Published : 01 Sep 2015
In this work, using quantum mechanics, the interaction of drug carvedilol with (5, 5) COOH functionalized single wall carbon nanotubes (SWNT) have been studied. All of the calculations have been performed using a hybrid density functional method (B3LYP) in gas and solution phases. Two possible modes of covalent interaction of carvedilol onto COOH functionalized SWNT were investigated. Quantum molecular descriptors and frontier orbital analysis in the drug-nanotube systems were studied. It was found that bonding of carvedilol to COOH functionalized carbon nanotubes through hydroxyl group is stronger than amino group.
KEYWORDS:Carvedilol; COOH Functionalized Carbon Nanotubes; Drug Delivery; Quantum Molecular Descriptors
Download this article as:| Copy the following to cite this article: Rahbar M, Morsali A, Bozorgmehr M. R, Beyramabadi S. A. DFT study on the covalent adsorption of drug carvedilol onto COOH functionalized carbon nanotubes. Orient J Chem 2015;31(3). |
| Copy the following to cite this URL: Rahbar M, Morsali A, Bozorgmehr M. R, Beyramabadi S. A. DFT study on the covalent adsorption of drug carvedilol onto COOH functionalized carbon nanotubes. Orient J Chem 2015;31(3). Available from: http://www.orientjchem.org/?p=10531 |
Introduction
Nowadays, much attention has been paid to research on new and effective drug delivery systems. The object of the design of these systems is to improve the functioning of these drugs. Although toxicity and low solubility create limitations in the applicability of carbon nanotubes, but laboratory-wise, numerous reports concerning the use of carbon nanotubes as the carrier molecules for drugs have been presented. A major portion of researches in this field, at both theoretical and experimental levels, has been allocated to analysis of the possibility of adsorption of drugs onto the sidewall of carbon nanotubes without considerable dysfunction in its electronic structures1-4.
Also, interaction of carbon nanotubes with organic and aromatic molecules through covalent and noncovalent functionalization has been investigated. The main motive behind such investigations has been to improve their solubility and dispersability in aqueous and organic solvents5-9.
Carvedilol is used to treat heart failure and high blood pressure. It also is used to treat cancer10. Carvedilol is often used in combination with other medications. In spite of extensive use of carbon nanotubes in drug delivery, so far, molecular mechanism of adsorption of carvedilol in water by these nanotubes has not been investigated. In this work, using quantum mechanical methods, the covalent adsorption of carvedilol onto COOH functionalized carbon nanotubes was studied.
Computational Details
All of the present calculations have been performed with the B3LYP11-13 hybrid density functional level and 6-31G(d,p) basis sets using the GAUSSIAN 03 package14.
The solvent has an important role in chemical reactions explicitly15-21 or implicitly. The implicit effects of the solvent was considered by using the polarized continuum model (PCM).22, 23 In the PCM method, the molecular cavity is made up of the union of interlocking atomic spheres.
All degrees of freedom for all geometries were optimized in solution (water) phase. The PCM calculations were performed on carvedilol, a pristine and COOH functionalized armchair (5,5) SWCNT comprising 90 atoms (10 Å). In optimization of the structures, no use has been made of approximations such as the ONIOM24 and in spite of high computational cost, we preferred the results obtained have a good accuracy.
Results and Disscussion
The optimized geometry of carvedilol (CAR) in solution phase is shown in Fig. 1. Carvedilol may interact with COOH of functionalized SWCNT (NTF) through amino and hyroxyl groups to form covalent bonds.
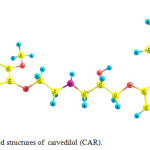 |
Figure 1: Optimized structures of carvedilol (CAR). Click here to View figure |
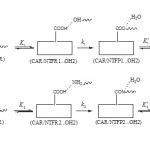 |
Scheme 1 Click here to View scheme |
Scheme 1 shows the mechanism of covalent adsorption of CAR onto COOH functionalized carbon nanotube where and are equilibrium constants and is rate constant. In the this mechanism, CAR/NTFR1 and CAR/NTFR2 are converted into the products CAR/NTFP1 ( pathway) and CAR/NTFP2 ( pathway) by losing H2O, respectively.
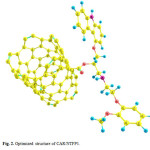 |
Figure 2: Optimized structure of CAR/NTFP1. Click here to View figure |
As shown in Scheme 1, this mechanism correspond to a substitution of OH from NTF by O (N) of CAR to give product CAR/NTFP1 (CAR/NTFP2)
These two configurations are depicted in Figs. 2 and 3. The absolute energies of CAR, CAR/NTFP1 and CAR/NTFP2 are shown in Table 1. Among the two configurations of CAR/NTFP1 and 2, the first one has more negative energy and more satable by 78.59 kJ/mol.
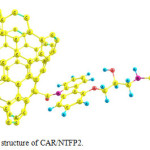 |
Figure 3: Optimized structure of CAR/NTFP2. Click here to View figure |
In describing stability and chemical reactivity of different systems, quantum molecular descriptors have been used like the chemical potential, the global hardness, the electrophilicity index and etc.
The chemical potential (μ) which shows escape tendency of an electron from equilibrium, is defined as follows:
μ = -(I+A)/2 (1)
where I= -EHOMO is the ionization potential and A= -ELUMO is the electron affinity of the molecule.
The global hardness (η) shows the resistance of one chemical species against the change in its electronic structure (Equation (2)). Increase in causes an increase in the stability and a decrease in reactivity.
η = (I-A) (2)
Electrophilicity index (ω) was defined by Parr as follows:25
ω = μ2 | 2η (3)
Tabel 1 represents the values of the quantum molecular descriptors calculated for different configurations in the solution phase. In this table, besides quantum molecular descriptors, Eg (HOMO-LUMO energy gap) was also presented. Eg notably shows a more stable system.
Table 1: Quantum molecular descriptors for optimized geometries of different species in solution phase.
|
CAR/NTFP2 |
CAR/NTFP1 |
CAR |
Species |
|
-4.67 |
-4.65 |
-5.47 |
|
|
-3.53 |
-3.50 |
-0.71 |
|
|
1.14 |
1.15 |
4.75 |
|
|
4.67 |
4.65 |
5.47 |
|
|
3.53 |
3.50 |
0.71 |
A |
|
0.57 |
0.57 |
2.37 |
|
|
-4.1 |
-4.07 |
-3.09 |
|
|
14.74 |
14.53 |
2.01 |
|
|
-4881.781205 |
-4881.811166 |
-1340.642213 |
E(Hartree) |
According to the data in Table 1, η , I, and Eg related to the carvedilol drug are higher than CAR/NTFP1 and CAR/NTFP2, showing the stability of the carvedilol decreases in the presence of NTF and its reactivity increases. Also, in confirmation of the previous issue, it is observed that μ of the carvedilol becomes more negative in the presence of NTF. ω of the carvedilol increases in the presence of NTF, showing that the carvedilol acts as electron acceptor.
Frontier orbital analysis is relatively useful in the determination of the chage localization/delocalization in a molecule and based thereon, we can obtain a perspective of the electronic charge distribution in a molecule. The localization of electron density in HOMO makes the position to be nucleophilic.
The HOMO and LUMO orbitals of carvedilol, pristine NT, CAR/NTFP1 and CAR/NTFP2 are illustrated in Figs. 4 and 5, repectively. In NT, the HOMO is localized throughout the CNT sidewall along the C–C bonds parallel to CNT axis, whereas the LUMO is localized along the C-C bonds perpendicular to the CNT axis. Covalent functionalization leads to significant perturbation of the charge distribution of the nanotube,modulating the frontier orbitals around the site of functionalization. In the covalent functionalization, reorientation of the frontier orbital contribution from the HOMO is noted with the charge delocalized around the functional unit of the otherwise inert nanotube.
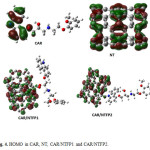 |
Figure 4: HOMO in CAR, NT, CAR/NTFP1 and CAR/NTFP2. Click here to View figure |
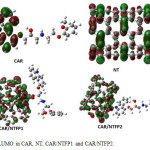 |
Figure 5: LUMO in CAR, NT, CAR/NTFP1 and CAR/NTFP2. Click here to View figure |
Conclusion
Using density functional theory, the effects of the adsorption of carvedilol onto COOH functionalized single wall carbon nanotubes (NTF) have been studied in detail in solvent environment. Two possible modes of covalent adsorption of carvedilol onto NTF were investigated. There are two possibilities for the formation of covalent bonds between carvedilol and COOH functionalized SWCNT, for the first possibility, carvedilol is bonded to NTF through hydroxyl groups and for the second one through amino groups . The product of first possibility is more stable.
Acknowledgment
We thank the theoretical research group in research center for animal development applied biology for allocation of computer time.
- Wong, B. S.; Yoong, S. L.; Jagusiak, A.; Panczyk, T.; Ho, H. K.; Ang, W. H.; Pastorin, G. Adv. drug deliv. rev. 2013, 65, 1964-2015.
- Sharifi, S.; Hashemi, M. M.; Mosslemin, M.; Mollaamin, F. J. Comput. Theor. Nanosci. 2014, 11, 1178-1183.
- Hosni, Z.; Bessrour, R.; Tangour, B. J. Comput. Theor. Nanosci. 2014, 11, 318-323.
- Saikia, N.; Deka, R. C. J. Mol. Model. 2013, 19, 215-226.
- Gad, E. A.; Al-Fahemi, J. H.; Khairou, K. S. J. Comput. Theor. Nanosci. 2014, 11, 404-408.
- Yahyaei, H.; Monajjemi, M.; Aghaie, H.; Zare, K. J. Comput. Theor. Nanosci. 2013, 10, 2332-2341.
- Naghsh, F. Orient. J. Chem. 2015, 31, 465-478.
- Yousefian, Z. Orient. J. Chem. 2014, 30, 1681-1693.
- Star, A.; Liu, Y.; Grant, K.; Ridvan, L.; Stoddart, J. F.; Steuerman, D. W.; Diehl, M. R.; Boukai, A.; Heath, J. R. Macromolecules 2003, 36, 553-560.
- Lin, C.-S.; Lin, W.-S.; Lin, C.-L.; Kao, C.-H. Int. J. Cardiol. 2015, 184, 9-13.
- Becke, A. D. Phys. Rev. A 1988, 38, 3098.
- Becke, A. D. J. Chem. Phys. 1993, 98, 5648-5652.
- Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785.
- Frisch, M.; Trucks, G.; Schlegel, H.;Scuseria, G.; Robb, M.;Cheeseman, J.; Montgomery, J.;Vreven, T.;Kudin, K.;Burant, J. Gaussian 03, Revision B.05 Gaussian Inc., Pittsburgh, PA,2003.
- Hooman Vahidi, S.; Morsali, A.; Ali Beyramabadi, S. Comput. Theor. Chem. 2012, 994, 41-46.
- Akbari, A.; Hoseinzade, F.; Morsali, A.; Ali Beyramabadi, S. Inorg. Chim. Acta 2013, 394, 423-429.
- Morsali, A.; Hoseinzade, F.; Akbari, A.; Beyramabadi, S. A.; Ghiasi, R. J. Solution Chem. 2013, 42, 1902-1911.
- Mohseni, S.; Bakavoli, M.; Morsali, A. Prog. React. Kin. Mec. 2014, 39, 89-102.
- Darzi, N.; Morsali, A.; Beyramabadi, S. A. Orient. J. Chem. 2015, 31, 1121-1125.
- Gharib, A.; Morsali, A.; Beyramabadi, S.; Chegini, H.; Ardabili, M. N. Prog. React. Kin. Mec. 2014, 39, 354-364.
- Morsali, A. Int. J. Chem. Kinet. 2015, 47, 73-81.
- Cammi, R.; Tomasi, J. J. Comput. Chem. 1995, 16, 1449-1458.
- Tomasi, J.; Persico, M. Chem. Rev. 1994, 94, 2027-2094.
- Dapprich, S.; Komáromi, I.; Byun, K. S.; Morokuma, K.; Frisch, M. J. J. Mol. Struct. (THEOCHEM) 1999, 461, 1-21.
- Parr, R. G.; Szentpaly, L. v.; Liu, S. J. Am. Chem. Soc. 1999, 121, 1922-1924.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


