Prediction of activity of carbonic anhydrase inhibitor drugs based on QSAR studies
Correspondence to: NaserDarzi (Email: n_darzi138@yahoo.com)
DOI : http://dx.doi.org/10.13005/ojc/310264
Download this article as:
![]()
A quantitative structure-activity relationship (QSAR) model, based on three quantum chemical descriptors obtained from the benzene sulphonamide derivatives using the density functional theory (DFT) method. Then this developed model was used to predict the benzene sulphonamide binding constant. The QSAR model has correlation coefficient R of 0.901 and the standard error of 0.646. Also, the predictive power of this model was further examined by leave-7-out cross validation procedure which the obtained statistical parameters were: Q2= 0.991 and SPRESS= 0.4686 that giving a good enough predictive power. The selected descriptorsare: molecular weight (MW), absolute hardness (AH), HOMO energy (HOMO), respectively.
KEYWORDS:Sulphonamides; Binding Constant; QSAR; DFT
Introduction
The carbonic anhydrases (CA) form a family of enzymes that catalyze the rapid interconversion of carbon dioxide and water to bicarbonate and protons (or vice versa), a reversible reaction that occurs relatively slowly in the absence of a catalyst. The active site of most carbonic anhydrases contains a zinc ion; they are therefore classified as metalloenzymes. One of the functions of the enzyme is to interconvert carbon dioxide and bicarbonate and in conclusion to maintain acid-base balance in blood, Stomachand other tissues, and also carbon dioxidetransmission of red blood cells and of red blood cells to tissues to facilitate the lungs. Change the function of this enzyme disrupts the body’s normal functions.
In between, Benzene sulphonamides important group of drugs that is capable of inhibiting carbonic anhydrase. Benzene sulphonamides are important groups of clinically drugs that be used in the treatment of diseases such as gastric ulcer, duodenal and intestinal disorders, glaucoma, infections and tumors are caused by high altitude (1-4). Also, additional products of them disarrange normal physiological functions (4).Whereas, considering the suitable does of drug and binding constant should be determined by experimental method that is limited (4) therefore, the development of theoretical model to predict the activities of these drugs are interesting and necessary. Quantitative structure activity relationship (QSAR) methods enable in prediction and interpretation of the activities of a wide range of organic and drug compounds based on the correlation between their activities and molecular characteristics (molecular descriptors). There are several reports about the applications of QSAR in this case.
In the probe, Kamal Kumar et al. revealed the relation of molecular size and inhabitation of CA by sufonamides.They developed the theoretical model based on plan of active sites of enzyme and demonstrated the nature of receptor-solfonamide joint (5). At the other investigation, Hansch et al. provided the QSAR model that consisted of formula for binding constant of benzene sulphonamides as the human carbonic anhydrase (6). Menziani et al. benefited the molecular mechanism Method for studying the interaction between 20 deprotonatedbenzenesulphonamides and carbonicanhydraseenzyme. Their results shown that the resection of enzyme-inhibitor is prevailed by the short-range van der Waals forces (7). Vijay et al. presented quantitative structure–activity studies on a group of sulphanilamide Schiff’s base inhibitors of carbonic anhydrase using distance-based topological indices. The regression analysis consequences indicated that the activities of the inhibiting carbonic anhydrase II (CAII) activity can be modeled excellently in multi parametric model (8). Gupta et al. performed a QSAR study on the inhibition of a few isomers of carbonic anhydrase and some metalloproteinases (MPs). They demonstrated well correlation with Kier’s first-order valence molecular connectivity index (1chi (v)) and electrotopological state indices of some atoms in the proposed model (9).Varma and Wagde,QSAR studies carried out on the basis of 50 substituted benzene sulfonamides. They investigated the effects of Balabanindexesin predicting activity of carbonic anhydrase inhibitor (10).Also,Felegari,Quantum chemical calculations performed another research based on DFT method for evaluation of anticancer drugs Cyclophosphamide.The results of this work revealed that quantum parameters affecting anticancer activity (11).
In the present work we try to predict the logarithm of binding constant of some sulphonamides derivatives from their quantum molecular descriptors by using multiple linear regression (MLR) techniques. For this purpose, we employed the density functional theory (DFT) method for geometrical optimization.
Experimental
The studied materials of this research work are 29 derivatives of benzene sulphonamide (12). Structure of these compounds and their experimental and predicted values of binding constant (log K) are available in table 1.Before anything else, the structures of all the derivatives of benzene sulphonamide have been optimized and then encoded the structural features (descriptors) of the molecules. We used of Gaussian (03) program (13) with B3lyp function (14, 15) and 6-311++G (d, p) basis set for geometrical optimization. Finally, quantum chemical descriptors were revealing for each molecule were obtained from the Gaussian output. Then the data set was separated into two groups: training and test sets. All molecules were placed by Y-ranking method in these sets. The training set, consisted of 22 molecules, was used for the model generation and the test set, consisted of 7 molecules, was used to take care of the over training and evaluate the prediction power of the generated model.
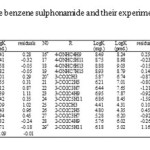 |
Table1: Structure of the benzene sulphonamide and their experimental and predicted values |
Descriptor selection and model development
For suitable descriptor selection, it should be considered the correlation between the calculated structural descriptors and the experimental binding constant values. Multiple linear regression is one of the most used modeling methods for this case. At first, for screening the calculated descriptors, highly correlated descriptors were removed and the descriptors with constant or almost constant values for all molecules were eliminated from the collection of descriptors. At the end of this step a total of 7 descriptors were reminded to further investigations. These descriptors are: Heat of Formation (HF), Molecular Weight (MW), Total Energy (TE), HOMO Energy (HOMO), LUMO Energy (LUMO), Absolute Hardness (AH) and Electronegativity (ELEC). In order to successive decision for developing QSAR model, it should be stopped adding descriptors to the model during the selection procedure. Therefore, we benefited from ‘break-point’ procedure to control the model expansion (16). In this technique, improvement of the statistical quality of the models is analyzed by plotting the squared correlation coefficient values of the obtained models versus the number of descriptors that were involved in each model. Thus, stepwise-MLR procedure was applied to the training setand multi linear regression equations of up to 7 descriptors were developed. In this procedure we employed SPSS (V. 17) (17). Then variations of square of correlation coefficient (R2) against the number of descriptors in the models according to break-point procedure were recorded and are shown in fig 1. Pursuant to this figure led to the conclusion that the best model had 3 descriptors. These descriptors were listed in table 2 whit their correlation matrix. As can be seen in this table there are not any high correlation between these descriptors.
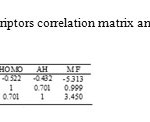 |
Table2: Descriptors correlation matrix and mean effects |
Results and Discussion
The best model was selected based on the statistics of correlation coefficient (R), standard error (SE) and Fisher-statistics value (F). Consequently, among different models the three-parameter model was chosen based on the break point procedure basis on the fig 1. A model result has been shown below.

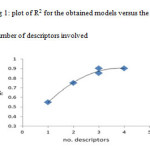 |
Figure1: plot of R2 for the obtained models versus the number of descriptors involved Click here to View figure |
The MLR predicted values of log K were shown in table 1. Finally, the leave 7-out cross-validation (L-7-O) was used to evaluate credibility and robustness of this model. For examining this test, the statistical parameters of Q2 and SPRESS were calculated (18).

In the above expressions, k is the number of independent variables (descriptors) in regression equation, n is the number of observations (data set), y– is the mean of dependent variable yΛi, is the predicted value and is experimental value of depended variable.
The resulted statistical values indicate that the proposed has stability, robustness, satisfactory fitting and prediction ability to predict the binding constant of benzene sulphonamide derivatives. We plotted the observed log K versus the calculated values of them in the fig 2. The excellent agreement between the experimental and calculated values of binding constant are demonstrate in this figure. The residuals of the MLR calculated values of their experimental values of benzene sulphonamide binding constant are plotted against their experimental values in fig 3. The random distribution of residuals about zero line verified that there is no systematically error in developed model.
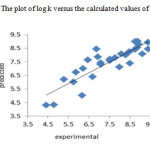 |
Figure2: The plot of log k versus the calculated values of them Click here to View figure |
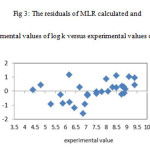 |
Figure3: The residuals of MLR calculated and experimental values of log k versus experimental values of them |
Also, we profited Y-scrambling test to investigate any chance correlation in our modeling (19). In this case, the dependent variable (log K) was randomly admixed and the original descriptors matrix is kept fixed then a new QSAR model was evolved. The results of this study are summarized in table 3. According to these results, it can be concluded that there was not any chance correlation between our data.
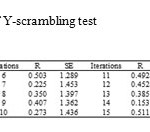 |
Table3: The results of Y-scrambling test Click here to View table |
Descriptor analysis and interpretation
By the explanation of descriptors of this model, obtaining the effected factors (MF) on the activity of benzensulphonamides is well possible. Here, a brief interpretation of these factors is given based on the results of mean effect analysis (20). This parameter calculated for each descriptor that was shown in table 2.

where; MFj is the mean effect for considered descriptor j, βj is the coefficient of descriptor j and dij is the value of interested descriptors for each molecule, n is the number of molecules in the data set and m is the number of descriptors in the model.
The value of MF manifests the relative importance of a descriptor in comparison with the other descriptors in the model. Also, the sign of descriptor represents the direction of variation in the values of activities resulted by increasing or decreasing the values of this descriptor. In this work, the results of mean effect analysis indicate that the order of importance of descriptors is: MW, AH and HOMO respectively.
MW with maximum mean effect is related to molecular size and is atom-type sensitive. The negative sign of this descriptor shows the inverse relation white log K (binding constant) (21). AH whit middle mean effect is an important quantum chemical descriptor that relates to energy levels and wave function of molecule and donates the stability of molecule as well as HOMO-LOMO energy gap. The positive mark of this descriptor shows the direct relation white log K (21). HOMO with minimum mean effect is related to information about reactivity-stability of specific region of the molecule. Its positive mark donates the direct relation white log K (21).
Conclusion
We have used quantum chemical descriptors basis of DFT calculations for QSAR studies of derivatives of benzene sulphonamides. The selected descriptors in proposed model can encode perfectly features of molecules which were responsible in benzene sulphonamide binding constant. Moreover, Q2 and SPRESS in leave-7-out cross-validation of MLR were used as a measurement of generalization performance. The obtained statistical parameters of this test demonstrate devised QSAR model is a simple, robust and significant tool to predict the activity of new sulphonamides drugs via main factors of structure and the quantum chemical indices.
References
- Bhat, M. A.; Imran, M.; Khan, S. A.; Siddiqui, N. Ind.J. pharmaceutical sci. 2005,67, 151-159.
- Siliveru, S.;Parthasarathy, T. Int. res.J. pharmacy. 2012,3, 213-215.
- Villar, R.;Encio, I.;Migliaccio, M.; Gil, M. J.; Martinez, M. V. Bioorga and Med Chem. 2004,12, 963-968.
- Supuran, T. C.;Scozzafava, A.;Casini, A. Med.Res.. Rev. 2003,23, 146-189.
- Kumar, K.;Bindal, M. C.; Singh, P.; Gupta, S. P. Int. J. Quant. Chem. 1981,20, 123-129.
- Hansch, C.;Mclarin, J.; Klein, J.;Langridge, R. Mol. Pharmacol. 1985,27, 493-498.
- Menziani, M. C.;Debenedetti, P. G.;Gago, F.; Richards, W. G. J. Med. Chem. 1989,32, 951-956.
- Agrawal, V. K.; Srivastava, R.;Khadikar, P. V.Bioorg. Med. Chem.2001,9, 3287-3293.
- Gupta, S. P.;Maheswaran, V.;Pande, V.; Kumar, D. J. Enzyme Inhib. Med. Chem. 2003,18, 7-13.
- Varma, R. G.; Wagde, r.Orient J. Chem. 2013, 29, 1621-1626.
- Felegari, Z. Orient J. Chem. 2014, 30, 1865-1875.
- Thakur, A.; Thakur, M.;Khadikar, P. V.;Supuran, C. T.;Sudele, P. Bioorg. Med. Chem. 2004,12, 789-793.
- Frisch, M.; Trucks, G.; Schlegel, H.;Scuseria, G.; Robb, M.;Cheeseman, J.; Montgomery, J.;Vreven, T.;Kudin, K.;Burant, J. Gaussian 03, Revision B.05Gaussian Inc., Pittsburgh, PA,2003.
- Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B. 1988,37, 785-789.
- Becke, A. D. J. Chem. Phys. 1993,98, 5648-5652.
- Fatemi, M. H.;Baher, E. Mol. Divers. 2009,13, 343-352.
- SPSS/PC, Statistical Package for IBMPC, Quiad software, Ontario, 1986.
- Fatemi, M. H.;Baher, E. SAR & QSAR Environ.Res. 2009,20, 77- 90.
- Fatemi, M. H.;Baher, E.;Ghorbanzade’h, M. J. Sep. Sci. 2009,32, 1-10.
- Fatemi, M. H.;Gharaghani, S.;Mohammadkhani, S.;Rezaie, Z.ElectrochimicaActa. 2008,53, 4276-4282.
- Tadeschini R. and Consonni V., Handbook of Molecular Descriptors,Wiley-VCH.Weinheim, (2000).








![]()




{kind=link}