Density functional theory, natural bond orbital and atoms in molecule analyses on the hydrogen bonding interactions in 2-chloroaniline – Carboxylic acid complexes
DOI : http://dx.doi.org/10.13005/ojc/310233
Download this article as:
![]()
In this study complexes formed via hydrogen bond interactions between 2-chloroaniline with carboxylic acids have been studied by density functional theory (DFT) using 6-311++G (d, p) atomic basis set. The relevant geometries, energies, and IR characteristics of hydrogen bonds have been systematically investigated. Thirteen 2-chloroaniline - carboxylic acid complexes were found involving N-H…N, H-O…H, N-H---O, O-H…N hydrogen bonds. The quantum theory of atoms in molecule (AIM) and natural bond orbital (NBO) have also been applied to understand the nature and strength of the hydrogen bonding interactions in complexes. Good correlations have been established between hydrogen bond lengths versus AIM topological parameter like electron density ( ) and its Laplacian ( ) at the bond critical points. The strength of the hydrogen bond between 2-chloroaniline – carboxylic acid complexes has been explored by calculation of stabilization energy (E (2)) between proton acceptor and proton donor under NBO analysis.
KEYWORDS:carboxylic acids; hydrogen bond; atoms in molecule (AIM); natural bond orbital (NBO)
Introduction
Hydrogen bonding is one of the most important type of interactions. It is used in determining properties and functioning of both natural and artificial systems from water to advanced materials and also living organisms1-6. The hydrogen bonding ability of different functional groups is also used as a tool in the selective binding of drugs to their targets. In addition to increase the interaction between or within molecules, formation hydrogen bonds can influence the reactivity of different active sites or can even change the reaction mechanism of chemical or biochemical processes7-12. The characterization of the hydrogen bond has given rise to a large number of experimental works as well as theoretical investigations in order to understand the energetics, the structures and the bonding properties of the complexes.
The main objective of this study is to reveal the nature of the hydrogen bonding interactions between –NH2 group of 2-chloroaniline and –COOH group of carboxylic acids which is explained theoretically. The investigation on hydrogen bonding interaction between 2-chloroaniline and carboxylic acid molecules may provide useful information concerning the bond strength and bond characteristics of compounds that are of fundamental importance for the radical chemistry and the synthesis of organic materials.
Hydrogen bond formation between amines and carboxylic acids is generally promoted by the use of stoichiometric coupling reagents such as carbodiimides. 2-chloroaniline compounds are considered import in various fields such as Agriculture, Pharmaceuticals, Rubber chemicals, manufacturing of synthetic dyes, organic pigments and also used as a parent substance in the production of antioxidants. Carboxylic acids are used in ink, pesticides, cosmetics, plastics, and rubber as a chemical constituent. These organic compounds are also important in industrial application with carboxyl groups in the use of fatty acids in making soaps, detergents, and shampoos 13-17.
In the present study, the structural and electronic properties of hydrogen bonds (N-H…N, H-O…H, N-H—O, O-H…N) in 2-chloroaniline – carboxylic acid complexes have been studied using density functional theory method (DFT). AIM analyses based on Bader’s atoms in molecules theory 18 and NBO analyses 19 have been performed to confirm the existence of hydrogen bonding and to find the information about charge transfer between the molecules.
Computational Details
The Becke’s three parameter exact exchange functional (B3)20 combined with gradient-corrected correlation functional of Lee–Yang–Parr (LYP)21 of DFT method has been employed to optimize the geometries of monomer and 2-chloroaniline – carboxylic acid complexes. All the possible orientations of 2-chloroaniline with carboxylic acids have been fully optimized using the above level of theory at 6-311++G (d, p) basis set. Thirteen conformers corresponding to the minimum energy points have been obtained
As polarity of molecule has great influence on intermolecular hydrogen bonding and hydrogen bonded orbitals requires large space occupation. Thus, diffuse and polarization functions augmented split valence 6-311++G (d, p) basis set is used for better description of geometrical optimization and natural bonding orbital (NBO) analysis. All these calculations are performed using Gaussian 0922
The interaction energies of all dimers were determined from the energy difference between the dimer and monomers in their minimum energy configuration. The basis set superposition error (BSSE) was eliminated using Boys and Bernard’s counterpoise method 23.
Further to evaluate the strength of the intermolecular interactions, NBO analyses were performed at B3LYP/6-311++G (d, p) level. In order to analyse the properties of the Hydrogen bond interactions in complexes, the AIM analyses were carried out by using Multiwfn program. At bond critical point (BCP) of Hydrogen bonds a topological analysis of electron density ( p) and its Laplacian (▽2) help us to evaluate the nature of Hydrogen bond. Finally, the formation of hydrogen bond in the equimolar binary mixture systems is identified by the experimental FT-IR spectra recorded at room temperature (298.15 K).
Results and Discussion
Structure and Energy Analysis
To analyze the intermolecular hydrogen bonds between 2-chloroaniline and carboxylic acids it can be considered thirteen different associations based on the characteristics of the monomers. The optimized possible structures of all dimers have been computed at B3LYP/6- 311++G (d, p) level of theory and presented in Figure 1.The structural parameters of hydrogen bonds between 2-chloroaniline and carboxylic acid complexes were calculated with B3LYP/6-311G (d, p) basis set and presented in Table 1.
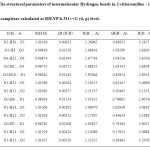 |
Table1: The structural parameters of intermolecular Hydrogen bonds in 2-chloroaniline – carboxylic acid complexes calculated at B3LYP/6-311++G (d, p) level. Click here to View table |
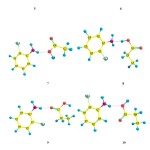 |
Figure1: Geometrical optimized structures using 6-311++G (d,p) of self associations 1 (2- Chloroaniline-2-Chloroaniline),2(Ethanoic acid-Ethanoic acid),3(Propanoic acid-propanoic), 4 (Butanoic acid-Butanoic acid) and of cross-associations 5,6,7 (2-Chloroaniline + Ethanoic acid) 8,9,10 (2-Chloroaniline + Propanoic acid),11,12,13 (2-Chloroaniline + Butanoic acid). Click here to View figure |
The molecular graphical analysis shows that different types of hydrogen bonds are formed in the obtained association between the donor (D-H) and acceptor group (A). The change of the D-H bond length during the process of the formation of hydrogen bond association can reflect the characteristic nature of H-bond. As shown in Table 1, all the values of ΔR(D-H) of hydrogen bonds are positive these values indicate that they are all red shifting hydrogen bonds. Furthermore a hydrogen bond parameter ΔR(D□□□Y)
24, is defined as ∆R (H…A) =R (H)vwr+ R(A)vwr ─ R(H….A) Where R(H)vwr and R(A)vwr are the Vander Waals radii of H and A acceptor atoms obtained by Bondi 25 respectively, R(D…A) is the distance of hydrogen-donor and hydrogen-acceptor.
During the process of the formation of D-H…A hydrogen bond electrons transfer happened between D- H and A group result in the shortening of H….A bond length. The shorter the H…A bond length is, then the stronger the interaction is, and vice versa. The H…A bond length of O4-H12…O1 (1.67744A0 in self-association) and O2-H10….N1 (1.92046 A0 in cross-association) hydrogen bonds are the shortest ones, which indicate that the strength of the two Hydrogen bonds should be the strongest.
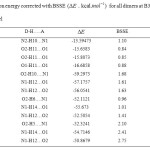 |
Table2: Interaction energy corrected with BSSE(ΔE. kcal.mol-1) for all dimers at B3LYP/6-311++G (d,p) level Click here to View table |
The ΔR(D□□□Y) can be estimated the strength of the hydrogen bond. From Table 1 the largest value of ΔR(H□□□Y) is 0.82954 A0 of the intermolecular OH…..N hydrogen bond in the cross-association 5 which represents the strongest hydrogen bond. The strength of the hydrogen bond in the self associations are presented in the order 3(O4-H12…O1)> 2(O2-H4…O3)> 4(O2-H1….O3)> 1(N2-H10…N1) and in the cross associations are in order 5(O2-H10….N1)> 8(O2-H6….N1)> 11(O2-H5….N1)> 9(N1-H14…O1)> 6(N1-H12…O1)> 12(N1-H14…O1)> 7(N1-H12…O2)> 10(N1-H12…O2)> 13(N1-H12…O2).
Interaction energy ( ΔE) for the hydrogen-bonded complex is calculated as the difference between the energy of hydrogen bonded complex and the summation of the energies of the each component monomers as given below:
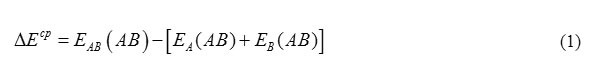
Where EAB optimized energy of hydrogen is bonded complex, EA (AB) and EA (AB) is the energies of the individual component monomers, respectively. Interaction energies are corrected for the basis set superposition error (BSSE) by virtue of counterpoise method 23. Hydrogen bonded complex is more stable if interaction energy is more negative compared to other hydrogen bonded complexes.
The counterpoise-corrected interaction energies, ΔEcp for all the dimers were computed at DFT, B3LYP with basis set 6-311++G (d, p). The calculated interaction energies summarized in Table 2 show that among the 13 complexes, the highest interaction energy is predicted for 3 (O2-H11…O1) in self-association with a value of -14.9573 and for 5 (O2-H10….N1) in cross-association with a value of -57.6173 . The trends in interaction energy are similar to the results of geometrical structures.
Electron Density Analysis
In the topological theory of AIM, when two neighboring atoms are chemically bonded, a bond critical point appears between them. The nature of chemical bonds and molecular reactivity are described by total electronic density p(r), Laplacian electronic density ▽2P(r) and the electronic energy density (H), which is composed by the electronic kinetic energy density (G) and the electronic potential energy density (V).
Topological analysis of electron density for the hydrogen bonds 26, 27 would be an easy to analyze the nature of hydrogen bonds formed in 2-chloroaniline and carboxylic acids complexes. The interactions are studied by considering the values of the electron density (p(r) ) and Laplacian of the electron density (▽2P(r)) at the bond critical points (BCP) of the N-H…N, H-O…H, N-H—O, O-H…N hydrogen bonds. The values of AIM topological parameters of the intermolecular hydrogen bond for all dimers are shown in Table
In the present study, the values of p(r) and ▽2P(r) varies from 0.011 to 0.047 a.u and 0.040 to 0.161 a.u. Maximum electron density in the self-association is 0.04702 a.u and in cross-association is 0.03389 a.u shows strong hydrogen bonds are observed for O-H….N interaction with high stability.
The positive values of Laplacian charge density in Table 3 for all molecules at BCPs are positive which reveal that electronic charges are depleted in the interatomic path, which is characteristic of closed shell interactions such as hydrogen bonds. The correlation between hydrogen bond length and electron density are inverse to each other i.e. increase in the hydrogen bond length corresponds to decrease in the electron density. Since there is increase in the distance results in reduced orbital overlap, electron density decreases along the bond. It is also observed that the Laplacian electron density and hydrogen bond lengths are also inversely related. The curves corresponding to correction coefficient for electron density and its Laplacian electron density with hydrogen bond length are shown in the Figure 2.The correlation coefficient of electron density, Laplacian electron density with hydrogen bond distance is observed to be 0.953 and 0.9144.
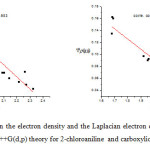 |
Figure2: The correlation between the electron density and the Laplacian electron density at BCP and hydrogen bond length at B3LYP/6-311++G(d,p) theory for 2-chloroaniline and carboxylic acid complexes Click here to View figure |
The total electronic energy density of the charge distribution may be expressed as

For strong hydrogen-bonding interaction, the total electronic energy density H is found to be negative (Table 3), showing partially covalent and partially electrostatic in nature, whereas this quantity is positive for medium and weak hydrogen bonds, revealing only electrostatic interactions for this bonding28.
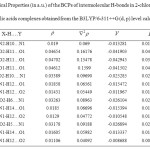 |
Table3: Topological Properties (in a.u.) of the BCPs of intermolecular H-bonds in 2-chloroaniline and carboxylic acids complexes obtained from the B3LYP/6-311++G (d, p) level calculations. Click here to View table |
Natural Bond Orbital Analysis
The natural bonding orbital (NBO) analysis has been a reliable tool for the rationalization of hydrogen bonds that well correlate with changes in bond length in accordance with the basic chemical concepts. It is also used to derive information on the changes of charge densities in proton donor and acceptor as well as in the bonding and antibonding orbitals. For each donor and acceptor, the stabilization energy E (2) associated with hydrogen bonding between sites i and j are given below29,
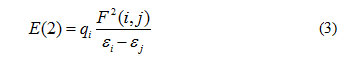
Where qi is the ith donor orbital frequency, εi , εj are diagonal elements associated with NBO Fock matrix. The stabilization energy between lone pair electrons (n) of the proton acceptor and antibonding orbitals (σ*) of the proton donor have been examined for various intermolecular hydrogen bonded complexes 29.
The NBO analysis has been performed here using DFT method for further probe to investigate the relative strength of all hydrogen bonded dimers. The oxygen/nitrogen atom with lone pair acts as donor and X-H(X=N, O) as acceptor in the strong intermolecular charge transfer interaction. The stabilization energies (E (2)) of intermolecular interactions of all dimers were performed by using second order perturbation theory. The stabilization energies between lone pair electrons of proton acceptor and antibonding orbitals of the proton donor have been examined for various intermolecular hydrogen bonds of self-association and cross association at B3LYP/6-311++G(d,p) level theory and tabulated in Table 4.
 |
Table4: Second-perturbation energies (E(2) / kJ·mol-1) of hydrogen bonds for all hydrogen bond associations obtained by NBO analysis at the B3LYP/6-311++G (d, p) level Click here to View table |
It is keen to note the point that there is a correlation between hydrogen bond length and stabilization energy E (2), i.e. shorter the bond length (strong hydrogen bond) larger the stabilization energy 30. In the case of self- association stabilization energy is more in 3 (20.63 kj.mol-1 ) and in the case of cross-association the stabilization energy is more in 5 (15.64 kj.mol-1 ) which coincides with the compared results of the interaction energies.
FT-IR Analysis
In order to examine the presence of hydrogen bond interaction between N-H and O-H groups of the both the systems, the infrared spectra are recorded at room temperature (298.15 K). Observing the experimental FT-IR spectra for the equimolar binary mixture of 2-chloroaniline+Ethanoic acid , there is a shift of 20 cm-1 wave number in the position of –NH and 16 cm-1 wave number in the position of –OH for the mixture compared with their respective pure spectrums (Fig. 3). The FT-IR spectra for the equimolar binary mixture 2-chloroaniline+Propanoic acid there is a shift of 16 cm-1 wave number in the position of –NH and 6 cm-1 wave number in the position of –OH for the mixture compared with their respective pure spectrums (Fig. 4). Similarly, in the FT-IR spectra for the equimolar binary mixture 2-chloroaniline+butanoic acid there is a shift of 12 cm-1 wave number in the position of –NH and 8 cm-1 wave number in the position of –OH for the mixture compared with their respective pure spectrums (Fig. 5) .These shifts are caused by the strong intermolecular interactions like hydrogen bonding between the O-H group of carboxylic acids (ethanoic acid, propanoic acid and butanoic acid) and N-H group of 2-chloroaniline. Thus, the FT-IR analysis convinces intermolecular hydrogen bonding of the equimolar binary mixture in both the systems effectively with proportionate variations in stretching wave numbers of –NH and –OH compared to their respective pure components 31.
 |
Figure4: Infrared spectra of pure 2-chloroaniline, pure Ethanoic acid and equimolar binary mixture of 2-chloroaniline + Ethanoic acid. Click here to View figure |
 |
Figure5: Infrared spectra of pure 2-chloroaniline, pure propanoic acid and equimolar binary mixture of 2-chloroaniline + Propanoic acid. Click here to View figure |
 |
Figure6: Infrared spectra of pure 2-chloroaniline, pure butanoic acid and equimolar binary mixture of 2-chloroaniline + Butanoic acid. Click here to View figure |
Conclusions
In this study it has been investigated that hydrogen bond interaction between 2-chloroaniline and carboxylic acid complexes using density functional theory at basis set of 6-311++G (d, p). The study reveals that
Optimized geometries and interaction energies reveals that, among all the dimers considered, 3 (O2-H11…O1) is found to be the most stable one in self-association and 5 (O2-H10….N1) is most stable in cross-association. The topological parameters calculated using the atoms in molecule methodology gives a proper explanation for the stability of the structure. The electron density and its Laplacian at BCPs correlate well with the hydrogen bond length. It is concluded that the stabilization energy E (2) calculated in NBO method indicates O-H…N hydrogen bonds are stronger among all the dimers.
Acknowledgement
The authors are thankful to Dr. M. Venkataraman, principal of Vignan Institute of Technology & Science, Hyderabad for providing the research facilities. They are also very thankful to Mr.K.Chandrasekhar Reddy, SSBN College, Anantapur, for providing Gaussian-09 facility and C-DAC, PUNE, India for providing the computational work.
References
- Affsprung, H. E.; Findenegg, G. H.; Kohler, F. J. Chem. SOC. 1968, 1364-1370.
- Kohler, F. Monatsh. Chem. 1969,100, 1151-1183.
- Becker, G.; Kohler, F. Monatsh. Chem. 1972,103, 556-570.
- Kohler, F.; Findenegg, G. H. J. Phys. Chem. 1974, 78, 1709-1714.
- Kohler, F.; Ber. Bumenges. Phys. Chem. 1978, 82, 576-582.
- Bobik, M.; Kohler, F.; Heger, G.; Lischka, H.; Schuster, P. Chem. Phys.Lett., 1976, 40, 66-71.
- Smith,D.A.; Wallwork, M.L.; Zhang, J.; Kirkham, J.; Robinson, C.; Marsh, A.; Wong, M. J Phys Chem B, 2000, 104, 8862-8870.
- Kokkoli, E.; Zukoski, C. F. Langmuir, 2001, 17,369-376.
- Boubour, E.; Lennox, R. B. Langmuir, 2000, 16, 4222-4228.
- Sugihara, K.; Shimazu, K.; Uosaki, K. Langmuir, 2000, 16, 4413-4415.
- Franco, M.; Nealy, P. F.; Campbell, S.; Teixeria, A. I.; Murphy, C. J. J. Biomed Mater Res, 2000, 52,261-269.
- Chapman, R.G.; Ostuni, E.; Yan, L.; Whitesides, G.M. Langmuir, 2000, 16, 6927-6936.
- Sewnarain, R.; Raal, J.D.; Ramjugernath, D. J. Chem. Eng. Data. 2002, 47,603-607.
- Bruice P.Y. Organic Chemistry. Prentice Hall, New Jersey, (1998)
- Pereiro, A.B.; Rodriguez, A. J. Chem. Thermodyn. 2008, 40, 1282-1289.
- Toumi, A.; Bouanz, M. J. Mol. Liq. 2008, 139, 55-60.
- Abdulagatov, I.M.; Tekin, A.; Safarov, J.; Shahverdiyev, A; Hassel, E. Int. J Thermophys. 2008, 29, 505-533.
- R.F.W. Bader, Atoms in Molecules. A Quantum Theory, Clarendon press, Oxford, UK, 1990.
- E.D. Gledening, A.E. Reed, J.A. Carpenter, F. Weinhold, NBO Version 6.
- Becke, A. D. Phys Rev A 1988, 38, 3098.
- Lee, C.; Yang, W.; Parr, R. G. Phys Rev B 1988, 37, 785.
- Frisch, M.; Gaussian 09 (Revisions A. 02). 2009, Gaussian, Inc., Walling ford CT, USA
- Boys, S.F.; Bernardi, F.; Mol. Phys. 1970, 19, 553–566.
- Chang, Guo.; Hui, Fang.; Rong-Yi, Huang.; Heng, Xu.; Gen-Hua, Wu.; Shi-Yong, Ye.; Chem. Phys. Letters 2013, 588, 97-101.
- Bondi, A.; J. Phys. Chem. 1964, 68, 441-451.
- M. Karthika, K. Senthil Kumar, R. Kanakaraju, Hydrogen bond interactions in hydrated acetylsalicylic acid, Comput. Theor. Chem. 2011.,966 ,167–179.
- L. Senthilkumar, T.K. Ghanty, S.K. Ghosh, Electron density and energy decomposition analysis in hydrogen-bonded complexes of azabenzenes with water, acetamide, and thioacetamide, J. Phys. Chem. A 2005.,109., 7575–7582.
- Zhao, Jun.; Khalizov, Alexei.; Zhang, Renyi. J. Phys. Chem. A, 2009, 113, 680-689
- Reed, A.E.; Curtiss, L. A.; Weinhold, F. Chem Rev 1988, 88, 899.
- Pal. S.; Kundu, T. K. ISRN Physical Chemistry . 2012, 2012, 1-12
- Silverstein, R. M.; Bassler, G. C.; Morrik, T. C. Spectroscopic Identification of Organic Compounds, 5th ed, (John Wiley&sons, Inc,), Singapore (1991).







ISSN Online: 2231-5039

![]()




{kind=link}