An eco-friendly and highly efficient route for N-acylation under catalyst-free conditions.
Laboratory of applied organic chemistry, Bioorganic Chemistry Group, Sciences Faculty, Chemistry Department, Badji Mokhtar-Annaba University, Box 12, Annaba, 23000. Algeria.
E-mail: noureddine.aouf@univ-annaba.dz
DOI : http://dx.doi.org/10.13005/ojc/310235
Download this article as:
![]()
An eco-friendly, simple, mild, chemo selective and highly efficient procedure for the acylation of primary and secondary amine function in various structurally and electronically aliphatic and aromatic compounds affording their corresponding N-Ac derivatives is developed. Mild conditions, simplicity and easier work-up are the main advantages of this method.
KEYWORDS:N-Acylation; amine; eco-friendly; protecting groups
Introduction
In the last two decades, the development of mild, non-toxic, low cost, eco-friendly methodologies has received much attention in modern organic transformations.1
The acylation of amines is fundamental chemical reaction for the production of important precursors, fine chemicals and pharmaceutical.2 The N-acylation of amines also provides an efficient and low-cost means to protect their -NH groups in a multi-step organic proccess.3,4 It is, generally, achieved by the reaction between amines and reagents containing acyl group. In the past few years, several methodologies were developed for N-acylation. This reaction is usually carried out with acylating reagent, such as acetic anhydride or acetyl chloride in the presence of acidic or basic catalysts in an organic medium. Recently, many methods were reported for on N-acylation of various structurally amines. The use of catalysts such as KF-Al2O3,5 ZnO,6 sodium formate,7 Amberlite IR120,8 FeCl3,9 Al(OTf)3,10 TiCl3(OTf),11 B(OCH2CF3)3,12 anhydrous NiCl2,13 iodine14 and homogeneous Lewis acid such as ZnCl215 have been reported for the N-acylation under various conditions. Also, microwave irradiation technique16, thermally decomposed Ni-Fe-hydrotalcite,17 heteropolyanion-based ionic liquids18 were used for the transformation of amines to their corresponding N-Ac derivatives. However, many of these methods suffered from various drawbacks such long reaction times, formation of side-products during base-catalyzed reactions.
As a consequence of serious pollution problems, the adoption of new methods for minimizing the negative impact and process optimization is an urgent priority19. In this context, the development of mild and eco-friendly N-acylation reaction continue to attach a great deal attention.20 An environmentally benign approach is described, where the N-Ac derivatives were prepared chemoselectively in water/micro-reaction system.21,22
In continuation of our interest toward the development of useful green synthetic procedure,23-28 we report the N-acylation of various structurally amine, amino alcohols and sulfonamides under catalyst-free conditions.
Results and Discussion
In order to find out the most effective reaction conditions, aniline was chosen as a model substrate. We first examined the influence of the solvent at room temperature. The reaction of aniline (1 mmol) with acetic anhydride (1.2 mmol) was studied using various solvents (THF, CH2Cl2, CHCl3, Et2O, EtOAc and H2O) at room temperature (Table 1). As shown in Table 1: entries 01-07, the reaction was completed within 5-15 min, the acylated product was obtained in good to excellent yields and the nature of solvent does not influenced the reaction.
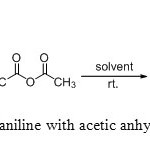 |
Scheme1: N-acylation of aniline with acetic anhydride in various solvents |
Table1: acylation of aniline in various solventsa
|
Entry |
Solvents |
Time (min) |
Yields (%)b |
|
01 |
THF |
6 |
75 |
|
02 |
CHCl3 |
5 |
79 |
|
03 |
CH2Cl2 |
5 |
81 |
|
04 |
Et2O |
10 |
76 |
|
05 |
EtOAc |
12 |
72 |
|
06 |
CH3CN |
7 |
78 |
|
07 |
H2O |
5 |
90 |
|
08 |
No solvent |
5 |
89 |
aReaction conditions: aniline (1mmolanydrid acetic (1mmol). bIsolated yield
For typical reaction, we achieved the reaction without solvent and got the similar yields. Also, time necessary for completion of the reaction was the same, thus we observed that the solvent does not have any role in this reaction.
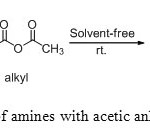 |
Scheme2: N-acylation of amines with acetic anhydride in solvents free. Click here to View scheme |
Encouraged by the preliminary result and to increase the scope of this reaction, we attempt this study to aliphatic, aromatic and heterocyclic amines (Scheme 2). The results are summarized in Table 2. In all cases, we obtained the N-acylated product with good to excellent yields. The best result was obtained with p-nitroaniline as substrate, affording N-acylated product in 91% yield after 8 minutes.
The results showed that all products (1-19) are not influenced by the electron-withdrawing and electron-releasing substituents.
 |
Table2: Acylation of amine Click here to View table |
In order to expand the scope of this new protocol, we investigated the acylation to a variety of aminoalcohols (alaninol, valinol, leucinol and phenylalaninol) (Scheme 3).
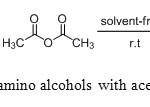 |
Scheme3: N-acylation of amino alcohols with acetic anhydride in solvents free. Click here to View scheme |
As it can be seen from results in Table 3, the reaction worked very well, and it was quite satisfactory considering that it was carried out without the use of any base, catalyst and solvent. The isolated yields of products (Table 3, Entries 20-23) were in the range of 85-90% after few minutes of reaction.
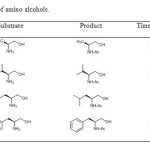 |
Table3: Acylation of amino alcohols. Click here to View table |
The simplicity and the chemoselectivity of this method can be determining of its application in the organic synthesis and particularly in peptide synthesis.
It is noteworthy that the reaction preserves the regioselctivity and stereochemical integrity of N-acyl amino alcohols, where the configuration of the chiral center was not affected by the conditions of reaction and the optically pure N-acyl derivatives were confirmed by optical rotation and comparison with literature.
The structures of all compounds were unambiguously confirmed by spectroscopic methods (IR, 1H, 13C NMR and elemental analysis). For the resulting compounds (Table 3, Entries 18-21), IR spectra showed bands at 1650-1700 cm-1(C=O) acetyl. 1H-NMR spectra showed the shift of proton corresponding to CH3 acetyl at 2.25 ppm.
To increase the scope of this reaction, we extended this study to sulfonamides29 (scheme 04).
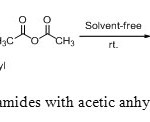 |
Scheme4: N-acylation of sulfonamides with acetic anhydride under solvents free conditions. Click here to View scheme |
Unfortunately moderate yields of the corresponding products ware formed with longer reaction times (table 04). Thus we thought that the sulfonamide require the use of a small amount of solvent.
For this reason and after referring to table 1, we decide to use the solvent who showed the best results; water. We treated sulfonamide (1mmol) in water with acetic anhydride in the absence of any catalyst (scheme 05). We choose N-benzyl sulfonamide as a model substrate. The reaction was complete in 8 minutes and expected product was obtained in (92%).
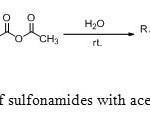 |
Scheme5: N-acylation of sulfonamides with acetic anhydride in water. Click here to View scheme |
 |
Table4: Acylation of sulfonamide. Click here to View table |
Conclusions
In summary, we developed new and efficient acylation of primary and secondary amines, aminoalcohols and sulfonamides. This new process has several advantages, such as high yield, short reaction times, low cost and simple experimental procedure. This method provides a green and much improved protocol over the existing methods.
Experimental Section
General
All commercial chemicals and solvents were used without further purification. Melting points were determined in open capillary tubes on a Bûchi apparatus and are uncorrected. 1H and 13C NMR spectra were recorded in a 250 MHz Brücker spectrometer. Chemical shifts are reported in (ppm) units with tetramethylsilane (TMS) as reference. All coupling constants (J) are reported in Hertz. Multiplicity is indicated as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), and combinations of these signals. All reactions were monitored by thin layer chromatography (TLC) on silica Merck 60 F254 percolated aluminium plates. Column chromatography was performed on Merck silica gel (230-400 mesh).
Typical experimental procedure for the synthesis of N-Acylated amines, amino alcohols and sulfonamides
In a 50mL round-bottomed flask, a mixture of amine or amino alcohol (1 mmol) and acetic anhydride (1.2 mmol) was stirred at room temperature for the appropriate time. After completion of the reaction, as monitored by TLC, the reaction mixture was dissolved in ether (5 mL) and was allowed to stand at room temperature for 1 hour. During this time, crystal of product formed, which were collected by filtration.
In the case of solid substrates (sulfonamides), the same protocol was used. However, the use of water was required for the solubility of the mixture. The N-acylated sulfonamides were collected by crystallization from diethyl ether.
Acknowledgements
This work was generously supported by the (Direction Générale de la Recherche Scientifique et du Développement Technologique, DGRS-DT), Algerian Ministry of Scientific Research, (FNR 2000). Fruitful discussions with Pr. Jacques Lebreton, Nantes University, France are gratefully appreciated.
References
- a Laboratory of applied organic chemistry, Bioorganic Chemistry Group, Sciences Faculty, Chemistry Department, Badji Mokhtar-Annaba University, Box 12, Annaba 23000. Algeria. Corresponding author: noureddine.aouf@univ-annaba.dz
- † Spectral data for N-Ac compounds prepared in this work are available in the supporting information joined to this manuscript.
- (a) Welton, T. Chem. Rev. 1999, 99, 2071; (b) J. S. Wilkes, Green Chem. 2002, 4, 73; (c) Zhang, Q.; Zhang, S.; Deng, Y. Green Chem. 2011, 13, 2619.
- (a) Hallettand, J. P.; Welton, T. Chem. Rev. 2011, 111, 3508; (b) Zhang, Q.; Zhang, S.; Deng, Y. Green Chem. 2011, 13, 2619. (c) Taylor, J. E.; Bull, S.D. In Comprehensive Organic Synthesis II, 2nd edn.; Elsevier Ltd. 2014; Vol 6, p 427.
- Greene, T. W., Wuts, P. G. M. In Protective Groups in Organic Synthesis; 3rd edn.; Wiley & Sons: New York; 2007.
- Kocieński, P. J. In Protecting Groups; Georg Thieme Verlag: New York, 1994.
- Mihara, M.; Ishino, Y.; Minakara, S.; Komatsu, M. Synthesis, 2003, 2317.
- Hosseini-Sarvari, M.; Sharghi, H. J. Org. Chem. 2006, 71, 6652.
- Brahmachari, G.; Laskar, S. Tetrahedron Lett. 2010, 51, 2319.
- Bhojegowd, M. R. M.; Nizam, A.; Pasha, M. A. Chin. J. Catal. 2010, 31, 518.
- Likhar, P. R. Journal of Molecular Catalysis A: Chemical, 2009, 302, 142.
- Kamal, A.; Khan, M. N. A.; Reddy, K. S.; Srikanth, Y. V. V.; Krishnaji, T. Tetrahedron Lett. 2007, 48, 3813.
- Firouzabadi, H.; Iranpoor, N.; Farahi, S.; J. Mol. Catal. A, 2008, 289, 61.
- Lanigan, R. M.; Starkov, P.; Sheppard, T. D. J. Org. Chem. 2013, 78, 4512.
- Meshram, G.; Patil, V. D. Synth. Commun. 2009, 39, 4384.
- Phukan, K.; Ganguly, M.; Devi, N. Synth. Commun. 2009, 39, 2694.
- Shekhar, A. C.; Kumar, A. R.; Sathaiah, G.; Paul, V. L.; Sridhar, M., Rao, P. S. Tetrahedron Lett. 2009, 50, 7099.
- Wang, X. -J.; Yang, Q.; Liu, F.; You, Q. -D.; Synth. Commun. 2008, 38, 1028.
- Choudhary, V. R.; Dumbre, D. K. Catalysis Communications, 2011, 12, 1351.
- Chen, Z.; Fu, R.; Chai, W.; Zheng, H.; Sun, S.; Qiang, L.; Yuan, R. Tetrahedron, 2014, 70, 2237.
- (a) Anastas, P. T.; Warner, J. C. Green Chemistry: Theory and Practice, Oxford University Press, 1988. (b) Gawande, M. B.; Bonifácio, V. D. B.; Luque, R.; Branco, P. S.; Varma, R. S. Chem. Soc. Rev. 2013, 42, 5522. (c) Gawande,M. B.; Bonifácio, V. D. B.; Luque, R.; Branco, P. S.; Varma, R. S. Chem Sus Chem 2014, 7, 24. (d) Matlack, A. S.; In Introduction to Green Chemistry, Marcel Dekker Inc., New York, 2001. (e) Lancester, M. In Green Chemistry: An Introductory Text, Royal Society of Chemistry, Cambridge, 2002. (f) Clark, J. H.; Macquarrie, D. J.; eds.; Handbook of Green Chemistry & Technology; Blackwell Publishers, Oxford, 2002. (g) Grieco, P. A. Eds.; In Organic Synthesis in Water; Blackie academic & professional; London, 1998.
- (a) Grieco, P. A. Eds.; Organic synthesis in water. London: Blackie academic & professional; London, 1998. (b) Pappas, K.; Zhang, X.; Tang, W.; Fang, S. Tetrahedron Lett. 2009, 50, 5741. (c) Chen, Z.; Fu, R.; Chai, W.; Zheng, H.; Sun, S.; Qiang, L.; Yuan, R. Tetrahedron, 2014, 70, 2237.
- Fruzsina, S.; Daniel, S.; Zoltan, N. RSC Adv. 2014, 4, 3883.
- Sato, M.; Matsushima, K.; Kawanami, H.; Yokoyama, T.; Ikuhsima, Y.; Maro Susuki, T. Lab Chip, 2009, 9, 2877.
- Cheraiet, Z.; Hessainia, S.; Ouarna, S.; Berredjem, M.; Aouf, N. -E. Green Chem. Lett. Rev. 2013, 6, 211.
- Amira, A.; K’tir, H.; Berredjem, M.; Aouf, N. -E. Monatch. Chem., 2014, 145, 509.
- Belghiche, R.; Cheraiet, Z.; Berredjem, M.; Abbessi, M.; Aouf, N.-E. Eur. J. Chem. 2012, 3, 305.
- Nadia. K.; Malika, B.; Nawel, K.; Belghit, M. Y.; Regaïnia, Z.; Aouf, N. E. J. Heterocycl. Chem. 2004, 41, 57.
- Lakrout, S.; K’tir, H.; Amira, A.; Berredjem, M.; Aouf, N. -E. RSC Adv., 2014, 4, 16027.
- K’tir, H.; Amira, A.; Berredjem, M.; Aouf, N. -E. Chem. Lett. 2014, 43, 851.
- (a) Berredjem, M.; Bouasla, R.; Aouf, N-E.; Barbey, C. X-Ray Structue Analysis Online. 2010, 26, 13. (b) Boufas, W.; Dupont, N.; Berredjem, M.; Berrezag, K.; Becheker, I.; Berredjem, H.; Aouf, N. -E. J. Mol. Struct.; 2014, 1074, 180.







ISSN Online: 2231-5039

![]()




{kind=link}