Catecholase activity investigations using in situ copper complexes continuing Schiff base derivatives with a theoretical calculation
A. Djedouani1,2, F. Abrigach3*, M. Khoutoul3, A. Mohamadou4, A. Bendaas5, R. Touzani3,6
1Laboratoire de Physicochimie Analytique et Cristallochimie des Matériaux Organométalliques et Biomoléculaires,Constantine 2Ecole normale supérieure de Constantine Algérie. 3Laboratoire de Chimie Appliquée et Environnement, LCAE-URAC18, COSTE,Faculté des Sciences, Université Mohamed Premier, BP524, 60000 Oujda, Morocco
4niversité de Reims Champagne-Ardenne, Institut de Chimie Moléculaire de Reims (ICMR), CNRS UMR 6229, Groupe de Chimie de Coordination, Moulin de la Housse, BP 1039, 51687 Reims Cedex2, France
5Université de Bordj Bou-Arréridj,El Anasser, Algérie.
6Faculté Pluridisciplinaire Nador BP 300, Selouane 62702 Nador, Morocco
DOI : http://dx.doi.org/10.13005/ojc/310110
Article Received on :
Article Accepted on :
Article Published : 19 Mar 2015
The study of catecholase activity of a series of Schiff base compounds using in situ copper complexes of 4-hydroxy-6-methyl-3-(1-(phenylimino)ethyl)-2H-pyran-2-one derivatives has been reported. The reaction rate depends on four parameters: The nature of the substitution in para position to the benzene ring, the nature of counter anion, the concentration of ligand and the nature of solvent. The highest rate activity is given by complex resulting from one equivalent of ligand L2 and two equivalents of copper acetate in methanol, which equal to 62.25 µmol.min-1.L-1.In other part, a theoretical study of such ligands using the semi-empirical method AM1 were also investigated. A good relationship founded between the maximal reaction rate (Vmax) and the HOMO energy (Pearson correlation: r=-0.794).
KEYWORDS:Schiff base; Catecholase; copper complexes; catalyst; semi-empirical method; AM1
Download this article as:| Copy the following to cite this article: Djedouani A, Abrigach F, Khoutoul M, Mohamadou A, Bendaas A, Touzani R. Catecholase activity investigations using in situ copper complexes continuing Schiff base derivatives with a theoretical calculation. Orient J Chem 2015;31(1). |
| Copy the following to cite this URL: Djedouani A, Abrigach F, Khoutoul M, Mohamadou A, Bendaas A, Touzani R. Catecholase activity investigations using in situ copper complexes continuing Schiff base derivatives with a theoretical calculation. Orient J Chem 2015;31(1). Available from: http://www.orientjchem.org/?p=7802 |
Introduction
Transition metal ions have an important role in biochemistry and biomimetic systems and may provide the basis of models for active sites of biological targets [1]. The presence of copper (II), iron (II) and zinc (II) is crucial in many biological processes. These metals are present in metallo-enzymes that involve the coordination of a metal ion as an active site and in which the metal plays the role of catalyst [2]. They also participate in the electron transport of O2, and the degradation of the superoxide ion O2− [3-5]. The coordination chemistry specially of dinuclear Cu(II) complexes has been developed very well with the goal of miming the active sites of the type 3 copper proteins/enzymes such as hemocyanin, tyrosinase and catechol oxidase. The oxidation of a wide range of o-diphenols to o-quinones is catalyzed by the catechol oxidases [6]. Thus, to understand more about this biological process and to search for new ligands that are able to contribute with other metals miming bio-organism puzzles, we examined a series of Schiff base compounds . These compounds have a wide range of applications, for example, catalyst, nonlinear optical materials, electroluminescent materials and others [7]. The synthesis of novel Schiff bases and the research of their physiological activities have become important due to their biological activities [8–10]. In continuation of our biometric work [11], we focused on the study of catecholase activity by using these ligands with different transition metals especially the copper via the oxidation of catechol to o-quinone.
Experimental
Chemistry of the studied ligands
All reagents were purchased from commercial sources and used without further purification. The ligands were prepared, and characterized according to literature [12], via an efficient condensation of an amine and dehydroacetic acid L0 in ethanol. The structures are shown in Scheme 1.
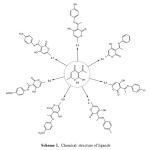 |
Scheme1: Chemical structure of ligands Click here to View Scheme |
Method of Catalytic Measurements
The measurements were made spectrophotometrically on UV–Vis spectrophotometer (in the COSTE: Centre de l’Oriental des Sciences et Technologies de l’Eau), following the appearance of o-quinone over time at 25 °C (390 nm absorbance maximum, ε = 1,600 L.mol-1.cm-1 in methanol [13]). The complexes were prepared in situ by successively mixing 0.30 mL of a solution (2×10-3 M) of CuX2, nH2O (X = Cl–, Br−, NO3−, CH3COO− or SO42−) with 0.15 mL of a solution (2×10-3 M) of ligand, then adding 2 mL of a solution of catechol at a concentration of 10-1 M. After that, the evolution of product absorbance was followed at 390 nm according to time after regulation in zero [11] (Scheme 2).
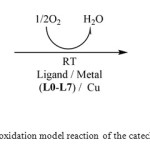 |
Scheme2: The oxidation model reaction of the catechol to o-quinone Click here to View scheme |
Results and Discussion
Catalytic Activity Studies
After realization of catechol oxidation reactions catalyzed by copper complexes with ligands L0–L7, the evolution of o-quinone absorbance was followed at 390 nm on the UV spectrometer UV–vis, we note that the catalytic activity varies from a complex to the other one, the obtained results for Cu(CH3COO)2 salt are summarized in below (Fig. 1)( more figures about the absorbance of o-quinone with different metal salts in the supplementary material) and the Vmax rates of the catechol oxidation were calculated in a time range of 5 min where the slope of the absorbance graph is maximal and collected in Table 1. In this part, we take one equivalent of ligand for two equivalents of metallic salt. In addition, the results obtained with L0 is not recorded here because it is too weak as if we were used the catechol alone.
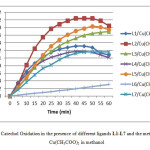 |
Figure1: Catechol Oxidation in the presence of different ligands L1-L7 and the metallic salt Cu(CH3COO)2 in methanol Click here to View figure |
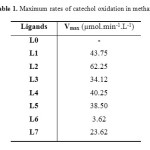 |
Table1: Maximum rates of catechol oxidation in methanol Click here to View table |
According to the obtained results, we notice that all copper complexes, formed in situ from ligands L1–L7 and copper salts (Cu(CH3COO)2 ,CuBr2,CuCl2, Cu(NO3)2,CuSO4) , catalyze the oxidation reaction of catechol to o-quinone, but with different rates which varying from 0.18 µmol. min-1.L-1 for the complex formed from ligand L6 and the metallic salt CuSO4 (weak catalyst) to 62.25 µmol.min-1.L-1 for the complex formed from ligand L2 and metallic salt Cu(CH3COO)2 (best catalyst). At this step, these results can be explained by the intervention of two factors: firstly, the nature of the core group on the para position of the benzene ring, in fact and in general , the donor group can increase the complexing properties of the ligand by the enrichment of the benzene ring, and promote stability in the complex. However, in presence of an electron attractor group, a disadvantage complex formation occurs by reducing the electron density on the sites of chelation and, therefore, reducing the rate of correspondence between the substrate and the complex. Secondly, the nature of counter-ion, so if it is strongly bonded to the metal as in the case of NO3−, Cl− and Br− ions, the binding of the substrate on the metal will be difficult, therefore reducing the catalytic power of the corresponding complex. Otherwise, the bonds formed between the metal and its counter-ion are not as strong (case of the CH3COO− ion), so the substrate can be replaced easily, which positively affects the rate of the reaction. In this part, we use different parameters to study the catalytic activity of the product L2 which gave the best results.
Effect of Ligand Concentration on the Catecholase Activity
In this study, the evolution of the oxidation of catechol to the o-quinone it’s followed in the presence of variable equivalents of ligand L2 and the metallic salt Cu(CH3COO)2. Three different concentration are used: One equivalent of L2 with one equivalent of Cu(CH3COO)2; Two equivalent of L2 with one equivalent of Cu (CH3COO)2 and One equivalent of L2 with two equivalent of Cu (CH3COO)2.
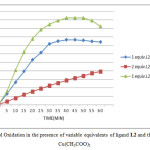 |
Figure2: Catechol Oxidation in the presence of variable equivalents of ligand L2 and the metallic salt Cu(CH3COO)2 Click here to View figure |
From the figure above, when the increasing of the concentration of the metal salt in first 35 min happens, the absorbance increase as will, after that, the absorbance decrease due to the precipitation of the a complex in the cuvette. Also the calculation of the oxidation rates, confirmed the absorbance values founded. From these observations, we can conclude, that the efficiency of our catalyst increase in presence of an excess of metal relative to the ligand, this result can be explained by the presence of two Cu(II) ions into the enzyme catecholase structure as site active, which granted firstly as an idea that our complex must contain two copper ion to in order to make its catalytic function happens in good way, and secondly as an idea about our model complexes which could be seen as functional or structural models for catechol oxidases or related copper containing enzymes.
Effect of the Acetate Salts
From the precedent results, what would be observed mainly is that the best results are showed with Cu (CH3COO)2. Therefore, for identify if the CH3COO− ion have an effect on the catalytic activity, we were used in this study different acetate salts with different metal (Cu, Zn, Ni and Mn).
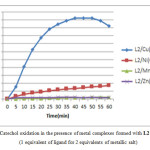 |
Figure3: Catechol oxidation in the presence of metal complexes formed with L2(1 equivalent of ligand for 2 equivalents of metallic salt)
|
The obtained results, showed very clearly that Cu (CH3COO)2 give the best results with a oxidation rate equal to 62.25 µmol.min-1.L-1, followed by Ni (CH3COO)2 with a value of 5.93 µmol. min-1.L-1, when the other salts give a very low rate < 1 µmol. min-1.L-1. As we can see here, the acetates has no effect on the oxidation rate in these conditions, but the difference between the rate values it is due to the metal ion which is very logical, because the reduction potential of (Cu 2+)(E0 =0.34 V) is more higher or favorable than the other metals tested, which make it more reactive than (Ni2+)(E0 =-0.25 V),(Zn2+)(E0 =-0.76 V) and (Mn2+)(E0 =-1.18 V).
Solvent Effect
The effect of the solvent on the oxidation reaction with ligand L2 and Cu (CH3COO)2 salt were studied in different solvents (MeOH, DMF, DMSO and CH3CN). As a simple comparison, we observe that the absorbance in MeOH is higher than in other solvents with a maximal value of 2.044 after 45 min of experience, before decreasing after that due to the precipitation of a complex in the cuvette, which is not the case in other solvents specially the DMF because this complex is very soluble in this solvents. So, the methanol is better solvent than the others for this reaction, and that can be explained by its protic and polar nature, because this kind of solvents are very known by their strong solvation of various anions X− (in our case X− = Cl−, Br− NO3−, CH3COO− and SO42−) through hydrogen bonds, which make them isolated and therefore are not very reactive in this type of solvents. The copper cations are also solvated by the electronegative pole of the solvent but is weaker than the solvation by a hydrogen bond, and became moderately reactive.
On the other hand, we observe that the positive charge is diffused, whereas the negative charge is concentrated on a single atom in the dipolar and aprotic solvents ( like DMF, DMSO,CH3CN,…) which mean that kind of solvents are very strongly solvating the (Cu2+) cations making them little or no reactive. For this, the formation of the copper complex is very difficult in this case, which negatively and indirectly affects the oxidation rate of the catechol to the o-quinone.
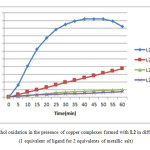 |
Figure4: Catechol oxidation in the presence of copper complexes formed with L2 in different solvents. (1 equivalent of ligand for 2 equivalents of metallic salt) Click here to View figure |
Substitution effect
The effect of substitution was studied for three ligands L7, L8 and L9 respectively substituted in Para, Meta and Ortho by a NH2 group (Scheme 3), the Cu (CH3COO)2 is used as metal salt and the methanol as solvent.
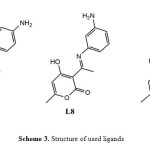 |
Scheme3: Structure of used ligands Click here to View Scheme |
 |
Figure5: Catechol oxidation in the presence of copper complexes formed with L7, L8 and L9. (1 equivalent of ligand for 2 equivalents of metallic salt) Click here to View figure |
 |
Table2: Oxidation rate of catechol oxidation in methanol
|
Comparing the unsubstituted ligand L2 with L7, L8, and L9, which have the same molecular formula, we note clearly that the existence of a substituted group on the benzene ring play an important role in the decrease of the catalytic activity of our compounds. As we can see from the Fig. 5 and the Table 2 the ligands substituted in para and meta position gives the best result with Cu (CH3COO)2 with a rate of 23.62 and 24.07 µmol.min-1.L-1 successively . For the two ligands L7 and L8, the shape of the curve of absorbance versus time is the same in the first 35 min, but after that, the absorbance in the presence of L7 remains fixed during the rest of the time unlike the ligand L8, which decreases due to the formation of a complex in the cuvette. However, the absorbance in the presence of L9 is weak probably due to the steric effect caused by the amino group, which prevents the (Cu2+) cations from complexing better with the ligand, which is not the case in the two other ligands, where the NH2 group is more far from the coordination site proposed.
Theoretical Study
In this part, we examined a theoretical study to find a correlation between our experimental values and the calculated values obtained by the quantum mechanical calculations [14, 15] of the Vmax (Table 3) and the molecular orbitals energy HOMO and LUMO.
Table 3. Oxidation rate of catechol oxidation in presence of the ligand and the copper metallic salt Cu(CH3COO)2
|
Ligands |
Vmax (µmol.min-1.L-1) |
|
L1 |
43.75 |
|
L2 |
62.25 |
|
L3 |
34.12 |
|
L4 |
40.25 |
|
L5 |
38.50 |
|
L6 |
3.62 |
|
L7 |
23.62 |
|
L8 |
24.07 |
|
L9 |
5.86 |
Method of Calculation
The semi-empirical methods are done on Hyperchem program version 8.0.6 for Windows [16] running on a Windows seven with a core i5 vPro. The initial geometry optimization of the studied molecules were performed with the molecular mechanics (MM+) force field [17, 18], where the lowest energy conformations are obtained. Further, Geometry optimization was done by performing the semi-empirical molecular orbital theory at the level parametric method (AM1) [19], within restricted Hartree–Fock (RHF) level [20]. The Polak–Ribier algorithm was used for the optimization [21]. The convergence is set to 0.01 kcal/mol.
Theoretical Calculations
The molecular orbitals energy: LUMO (lowest unoccupied molecular orbital) as an electron acceptor (EA) represents the ability to obtain an electron and HOMO (highest occupied molecular orbital) represents the ability to donate an electron (ED). The HOMO and LUMO energy gap explains the fact that eventual charge transfer interaction is taking place within the molecule. The values of the difference between the HOMO and LUMO orbitals (LUMO-HOMO), known as energy band gap (ΔE), are also given in Table 4. As it is shown in Fig. 6a and Fig. 6b (spatial distribution of the HOMO and LUMO of all molecules is presented in the supplementary material (Fig. 6c and Fig. 6d)), the HOMO orbitals are localized on the benzene ring and the imine function, distribution of molecular orbitals of LUMO is localized on pyrane ring.
 |
Table4: Molecular orbitals energies of ligands L0-L9 calculated with AM1 Click here to View table |
 |
Figure6a: The HOMO orbitals of L1 Click here to View figure |
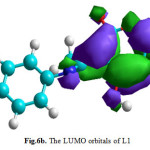 |
Figure6b: The LUMO orbitals of L1 Click here to View figure |
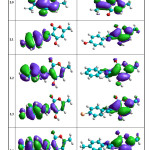 |
Figure 6 C Click here to View figure |
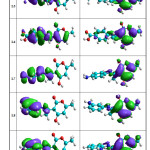 |
Figure 6D Click here to View figure |
Correlation
A study was conducted using IBM SPSS Statistics v 21 to find a correlation between the maximal reaction rate (Vmax) and the HOMO energy. The results were given in Table 5 and Fig. 7. Correlation quantifies the extent to which two quantitative variables, here Vmax and EHOMO. When high values of Vmax are associated with high values of EHOMO a positive correlation exists. However, a negative correlation obtained, when high values of Vmax are associated with low values of EHOMO.
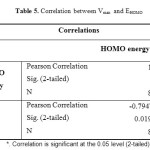 |
Table5: Correlation between Vmax and EHOMO Click here to View table |
A strong correlation is observed between the maximal reaction rate (Vmax) and HOMO energy, so the correlation is significant (r = -0.794; P = 0.019), here the correlation is negative.
 |
Figure7: Dependence of Vmax to EHOMO Click here to View figure |
 |
Figure 7B Click here to View figure |
These results show the good linear correlation between the maximal reaction rate and HOMO energy. This correlation proves the existence of a relationship between the catalytic activity, which is presented by Vmax, and HOMO energy, so we can estimate Vmax by study of the HOMO energy for this kind of ligands.
Conclusion
In conclusion, we have found that our compounds L1–L7 present a variant capacity to catalyze the oxidation reaction of the catechol to o-quinone at ambient conditions using the oxygen of atmosphere as oxidant. The catalytic activity of these complexes formed in situ is influenced by the nature of counter anion in metallic salt, ligand and the metallic salt concentration, nature of solvent and the nature of substitute group on the benzene ring. In addition, the Quantum mechanical and statistical calculations showed a good correlation between the maximal reaction rate and the HOMO energy witch proves the existence of a strong relationship between the catalytic activity and the HOMO energy. It seems interesting to continue this work, firstly, on the synthesis of these complexes and test their reactivity as metallo-enzymes in the field of biomimetic catalysis. Secondly, trying to find a general Model of Cu:O2 intermediates and understanding their action mechanism in the oxidation reaction of the catechol to o-quinone.
Acknowledgement
Our thanks to the Mohamed First University for its financial support.
References
- M.V. Angelusiu, S.F. Barbuceanu, C. Draghici, G. L. Almajan, Eur. J. of Med.Chem. 2010, 45(5), 2055-2062.
- R. Saddik, F. Abrigach, N. Benchat, S. El Kadiri, B. Hammouti, R. Touzani, Res. on Chem .Inter.2012,38 (8), 1987.
- M.R. Smith, Biodegradation 1990, 1, 191.
- A.M. Orville, J.D. Lipscomb, Met. Ions Biol. Syst. 1992, 28, 243.
- M.A. Miller, J.D. Lipscomb, J. Biol. Chem.1996, 271, 5524.
- T. Klabunde, C. Eicken, J. C. Sacchettini, B. Krebs, Nature Struct. Biol. 1998, 5, 1084.
- U. Takafumi, Y. Norihiko, U. Masaki, M. Toshitaka, T. Yuichi, Y. Masako, PNAS 2006, 103 ,9416.
- Heine, G. DeSantis, J.G. Luz, M. Michael, W. Chi-Huey, Science 2001, 294 ,369.
- N. Rmmn, I.A. Kulandaisa, C. Thangaraja, Transmetal. Chem. 2003, 28, 29.
- G.W. Yang, X.P. Xia, C.X. Zhao, Chin, J. Appl. Chem. 1995,12, 13.
- (a). A. Zerrouki, R. Touzani, S. El Kadiri, Arab. J. Chem. 2011, 4, 459; (b). N. Boussalah, R. Touzani, I. Bouabdallah, S. El Kadiri, S. Ghalem, Int. J. Acad. Res. 2009, 2, 137; (c). M. El Kodadi, F. Malek, R. Touzani, A. Ramdani, Catal. Commun. 2008, 9, 966 ; (d). N. Boussalah, R. Touzani, I. Bouabbdallah, S. El Kadiri, S. Ghalem, J. Mol. Catal. A. 2009,306, 113 ; (e). A.Mouadili, A.Zerrouki, L.Herrag, B.Hammouti, S.El Kadiri, R. Touzani, Res. Chem. Intermed. 2012, 38, 2427–2433; (f). R. Saddik, M. Khoutoul, N. Benchat, B.Hammouti, S. El Kadiri, R. Touzani, Res. Chem. Intermed. 2012, 38,2457–2470; (g). A. Mouadili ,A. Attayibat , S. El Kadiri , S.Radi , R. Touzani, Applie Catalysis A: General 2013, 454, 93–99.
- S.Iguchi, Chem. Pharm. Bull. YOKYO 1964, 12, 318.
- O. Fadel, T. Chelfi, S. Abouricha, Z. Lamzira, N. Benchat, A. Asehraou, Rev. Microbiol. Ind. San.Environn. 2010, 4, 114.
- A.Zarrouk, B. Hammouti & R. Touzani, “DFT and Quantum Chemical Studies for Heterocyclic Compounds Used as Corrosion Organic Inhibitors” ,LAP Lambert Ac. Publishing Ed. ISBN 978-3-659-21601-5, 2012,Chaps 1-5, pages 89.
- (a). K. Bouhrira, A. Chetouani, D. Zerouali, B. Hammouti, A. Yahyia, A. Et-Touhami, R. Yahyaoui, R. Touzani, Res. on Chem .Inter.2014, 40 (2), 569-586; (b). I. Elouali, A. Chetouani, B. Hammouti, A. Aouniti, R. Touzani, S. El Kadiri, S.Nlate , Portugaliae Elect. Acta. 2013,31 (2), 53-78; (c). K. Bouhrira, A. Chetouani, D. Zerouali, B. Hammouti, A. Yahyia, A. Et-Touhami, R. Yahyaoui, R. Touzani, Res. on Chem .Inter. 2014, 40 (2), 569-586; (d). M. Kaddouri, M. Bouklah, S. Rekkab, R. Touzani, S.S. Al-Deyab, B. Hammouti, A. Aouniti, Z. Kabouche , Int. J. Electrochem. Sci.2012, 7 (9), 9004-9023; (e). N. Boussalah, B. Hammouti, S. El Kadiri, S. Ghalem, R. Touzani , Res. on Chem .Inter.2012.38, 2009-2023.
- Hyperchem 8.0.6. Molecular Visualization and Simulation Program Package, Hypercube Inc. Gainesville, 2007.
- U. Burkert, N.L. Allinger, Molecular Mechanics ACS Monograph, American Chemical Society, Washington, DC,1982, p. 177.
- J.-H. Li, S. Gallion, C. Bender, H. Wikstro¨m, N.L. Allinger, K.M. Flurchick, M.M. Teeter, J. Comput. Chem. 1989, 10, 503.
- M.J.S. Dewar, E.G. Zoebisch, E.F. Healy, J.J.P. Stewart, J. Am.Chem. Soc. 1985,107, 3902.
- C.C.J. Roothaan, Rev. Mod. Phys. 1951,23, 69.
- P. Fletcher, Practical Method of Optimization, Wiley, New York, 1990.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


