Synthesis of Precursor Imidazolium Salts for the Synthesis of N-Heterocyclic Carbines Used as Ligands for the Enantioselective Preparation of Heterosteroids Compounds
DOI : http://dx.doi.org/10.13005/ojc/300204
Download this article as:
![]()
The imidazolium salts 1-phenyl-3-(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)propyl)-1H-imidazol-3-ium bromide (4), and 1-(pyrimidin-2-yl)-3-(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)propyl)-1H-imidazol-3-ium bromide (5) were respectively prepared by coupling reaction of the precursors 1-phenyl-1H-imidazole (1) and 2-(1H-imidazol-1-yl)pyrimidine (2) with 2-(3-Bromopropyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (3) . The purity of these compounds was confirmed by spectroscopic data. The NMR spectra showed all the signals and the structures were confirmed by HRMS.
KEYWORDS:Imidazolium salts; ligand; NHC-Carnines; Heterosteroids
INTRODUCTION
Since the discovery of the first N-heterocyclic carbene (NHC) complexes in 1968 by Öfele[1] and by Wanzlick[2] and the isolation of the first stable free carbene in 1991 by Arduengo. A et al.,[3] NHCs are playing an increasingly important role in major areas of organometallic, organic, and polymer chemistry.[4] Their metal complexes are known as extremely versatile and stable catalysts for a wide range of reactions including C-C coupling reactions,[5-11] olefin metathesis,[12-15] hydroformylation,[16-17] and polymirization reactions.[18-19]
The control of the stereo selectivity in transition-metal-catalyzed organic transformation reactions depends on development of a versatile ligand that would strongly coordinate with the metal center[20]. Polydentate ligands having stereo directing groups could help control the stability and reactivity of mrtals efficiently in a variety of homogeneous catalysis reactions. Thus, a ligand that combines a strongly coordinating unit with a functional group having great influence on the electronic and steric properties of the metal center has considerable potential.
In recent years, there has been growing interest in using N-heterocyclic carbene (NHC) as a ligand for homogenous catalysis [21]. An attractive feature of NHC is not only its strong σ-donating capability to metals but also the possibility of varying the susbstituents on the nitrogen atom. It is possible that introduction of chiral substituents into NHC would result in enantioselective transformations in asymmetric catalysis. However, the standard chiral NHC obtained by this strategy often leads to low chiral inductions, mainly because of rapid internal rotation of the chiral substituents around the C-N axis. Therefore, to lock the N substituents in fixed conformation, a proposed strategy employs an NHC that bears both a chiral center and a hard chelating functional group on the N substituents resulting in generation of polydentate ligands [20.22].
In the last decade, heteroatom-functionalized NHC-metal complexes having stereodirecting groups have been developed. These are classified as NHC-based chelate ligands that incorporate a neutral or an anionic functional group. For the neutral functionalized NHC, a breakthrough has been achieved by Burgess, who reports highly efficient asymmetric hydrogenation catalysis based on carbene/oxazoline iridium complexes [23]. Other important chiral bidentate NHC complexes have been developed by Gade et al[24] and Douthwaite and coworkers [25] for enantioselective hydrosilylation and alkylation, respectively. For the anionic-functionalized NHC, anionic aryloxy- or alkoxo-tethered NHC has been designed and successfully applied in catalytic asymmetric transformations. Pioneering work was done by Hoveyda and coworkers [26], where metathesis and alkylation reactions proceeded with high enantioselectivity by the use of NHC-Ag complexes as ligand precursors. Arnold et al[27] as well as Mauduit and coworkers [28] independently introduced chelating alkoxy NHC-Cu compexes for asymmetric alkylations. Thus, the anionic, tightly coordinating polydentate NHC-ligand system is expected to enhance catalyst stability and to offer a key structure for the construction of efficient stereodirecting elements.
More recently, mixed systems, contaning one NHC and one pyrimidine coordinating site have been described as ligands for Ag(I)[30] and Hg(II)[30-31] complexes. Neither palladium nor platinum complexes of these ligands had been reported when we filed the patent[29] until very recently similar palladium complexes were published .[32-34] Related palladium complexes with nitrogen based heterocyclic ligands like NHC coupled pyridines, [35-46] bbenzimidazoles,[47] pyridazine,[48] and pyrazoles[49-50] have been described as ligands for transition metal complexes.
An important advance, Wang and Lin showed that Ag2O can be used to form a Ag-NHC complex from an imidazolium salt that readily transfers the NHC to palladium.[51] The great advantage of this method is the broad tolerance for sensitive N-substituents, which can be destroyed by conventional deprotonation of an imidazolium salt with strong bases. Transmetalation to various metal species gives a wide variety of NHCs coordinate to rhodium, copper, ruthenium, and iridium. However, Ag-induced oxidative degradation of the imidazolium precursors severely limits the use of this method.[52]
Herein, we present a nouvel series of mixed NHC-pyrimidine salts with alkyl, aryl and Lewis acid substituents. Moreover, the use of a Lewis acid could support the activation of an electrophile (aldehydes, imines,…) and confined the sphere of coordination by bringing closer the substrate to the catalytic center (metal) and to the chiral environment brought by the Lewis acid.
At present, the conception and the elaboration of new imidazolium salts for organometallic chemistry evolved towards a design with several functionalities to increase the affinity ligand/metal/substrate, so promoting, in most of the cases, bigger one reactivity and enantioslectivity in the catalytic process.
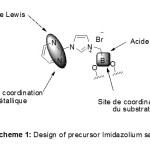 |
Scheme 1: Click here to View Scheme |
The design of our precursor imidazolium salts was so guided towards a multifunctional salt affording several complementary active sites for the metal and the substrate (scheme 1).
RESULTS AND DISCUSSION
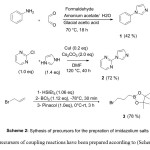 |
Schem 2: Click here to View Scheme |
The precursors of coupling reactions have been prepared according to (Scheme 2).
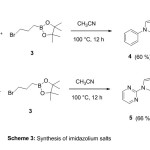 |
Scheme 3: Click here to View Scheme |
The precursors 1, 2 and 3 for the synthesis of imidazolium salts have been prepared according to the (Scheme 2).
We recently performed the synthesis of imidazolium salts 4 and 5 from the precursors which have been prepared previously by conversion of N-substituted imidazole 1 or 2 with 2-(3-Bromopropyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane 3 in CH3CN at 100 °C for 12h (Scheme 3).
The structure of compounds 4 and 5 was confirmed by NMR spectra.
EXPERIMENTAL
Synthesis of precursors of coupling reactions
Preparation of 1-phenyl-1H-imidazole (1)53
Glacial acetic acid (30.25 mL, 528.47 mmol, 4.3 eq), aqueous formaldehyde (9.04 mL, 328.14 mmol, 2.67 eq), and aqueous glyoxal (13.87 mL, 302.3 mmol, 2.46 eq) were transferred to a round bottom flask (150 mL) and heated at 70 °C. Asolution of glacial acetic acid (30.25 mL, 528.47 mmol, 4.3 eq), ammonium acatate in water (9.47 g/6.15 mL), and aniline (11.2 mL, 122.9 mmol, 1eq) was added drop-wise to the flask over a period of 1 hour. The solution was continuously stirred and heated at 70 °C for 18 h. The reaction mixture was then cooled to room temperature and added drope-wise to a stired solution of NaHCO3 (88.9 g) in water (900 mL), and the aqueous layer was extracted with diethyl ether (3×150 mL). The combined organic layers were washed with brine, dried over anhydrous MgSO4, and evaporated in vacuo The combined organic extracts were concentrated and the resulting residue was purified by column chromatography on silica gel (Petroleum ether/EtOAc = 20:80) to yield 1-phenyl-1H-imidazole as a yellow oil ( 7.48 g, 42 % yield).
1H NMR (300 MHz, CDCl3): δ = 7.20 (s,1H), 7.23-7.30 (m, 1H), 7.31-7.42 (m, 3H), 7.43-7.55 (m, 2H), 7.84 (s, 1H) ppm. 13C NMR (75 MHz, CDCl3): δ = 118.1, 121.4, 127.3, 129.8, 130.3, 135.5, 137.3 ppm.
HRMS (ESI) Calcd for C9H8N2 [M+H]+ : 145.0760. Found : 145.0760. C9H8N2 (144.17). Rf (AcOEt, 100 %) = 0.4 Flash chromatography eluent PE/AcOEt, 20/80. Yellow oil. Yield = 42 %.
Preparation of 2-(1H-imidazol-1-yl)pyrimidine (2)54
To a flame-dried Schlenk test tube with a magnetic stirring bar was charged with CuI (0.76 g, 4.0 mmol, 0.2 equiv), Cs2CO3 (13.00 g, 40.0 mmol, 2.0 equiv), imidazole (1.90 g, 28.0 mmol, 1.4 equiv), 2-chloropyrimidine (2.28 g, 20.0 mmol, 1.0 equiv) and DMF (40 mL) under argon. A rubber septum was replaced with a glass stopper, and the system was then evacuated twice and back filled with argon. The reactionmixture was stirred for 30 min at room temperature, and then heated at 120 °C for 40 hours. The reaction mixture was then cooled to ambient temperature, diluted with (20 mL) of ethyl acetate, filtred throught a plug of silica gel, and washed with ethyl acetate (100 mL). The combined organic extracts were concentrated and the resulting residue was purified by column chromatography on silica gel (Petroleum ether/EtOAc = 20:80) to yield 2-(1H-imidazol-1-yl)pyrimidine as a white solid (2.29 g, 78 % yield) m.p : 128-129 °C .
1H NMR (300 MHz, CDCl3): δ = 7.12 (s,1H), 7.16 (t, 3J = 4.9 Hz, 1H), 7.85 (t, 4J = 1.3 Hz, 1H), 8.58 (s, 1H), 8.65 (d, 3J = 4.9 Hz, 2H) ppm. 13C NMR (75 MHz, CDCl3): δ = 116.4, 118.8, 130.6, 136.0, 158.6 ppm.
Ms (ESI) for C7H6N4 [M+Na]+ : 168.7.
HRMS (ESI) Calcd for C7H6N4 [M+H]+ : 147.0665. Found : 147.0666.
Anal. Calcd for C7H6N4 (146.15) : C, 57.53; H, 4.4; N, 38.34 %.
Found : C, 57.61; H, 4.12; N, 38.09 %.
C7H6N4 (146.15). Rf (AcOEt, 100 %) = 0.4. Flash chromatography eluent PE/AcOEt, 20/80. White solid. Yield = 78 %.
Preparation of 2-(3-Bromopropyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane(3)55
To a mixture of allyl bromide (5.6 mL, 64.71 mmol, 1.0 equiv) and HSiEt3 (10.95 mL, 68.59 mmol, 1.06 equiv) was added a solution of BCl3 (1 M in hexane, 72.0 mL, 72.47 mmol, 1.12 equiv) at -78 °C under argon atmosphere. The resulting suspension was stirred at this temperature for 30 min, after which it was allowed to warm to room temperature for 1 hour. The mixture was cooled to 0 °C , and a solution of pinacol (7.64 g, 64.71 mmol, 1.0 equiv) in diethyl ether (30 mL) was added dropwise, and the mixture was stirred for an additional 3 hours. The solution was diluted with water (80 mL) and diethyl ether (40 mL), and the aqueous layer was extracted with diethyl ether (3×40 mL). The combined organic layers were washed with brine and dried with MgSO4 . After removal of the solvent under reduced pressure, the oily residue was purified by silica gel column chromatography (PE/EtOAc; 95/5) to yield 2-(3-bromopropyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane as a colorless oil. (12.61 g, 78 %)
1H NMR (400 MHz, CDCl3) δ (ppm) 0.74 (m, 2H), 1.18 (s, 12H), 1.89 (qt, 3J=7.7 Hz, 2H), 3.34 (t, 3J=6.9 Hz, 2H).
C9H18BBrO2 (248.90). Rf (PE/AcOEt, 80 /20) = 0.62. Flash chromatography eluent PE/AcOEt, 95/5. A colorless oil. Yield = 78 %.
Synthesis of imidazolium salts
Preparation of 1-phenyl-3-(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)propyl)-1H-imidazol-3-ium bromide (4)
To a 25 mL To a flame-dried Schlenk test tube with a magnetic stirring bar contaning 2-(3-Bromopropyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (3.0 g, 12.05 mmol, 1.0 eq) was added 1-phenyl-1H-imidazole (1.73 g, 12.05 mmol, 1.0 eq) and CH3CN (4 mL). T he mixture was then heated at 100 °C for 12 hours. The reaction mixture was then cooled to room temperature and diethyl ether (20 mL) was added to precipitate the imidazolum salt, filtred and washed with diethyl ether (20 mL). The solide was drieded under reduced pressure to yield1-phenyl-3-(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)propyl)-1H-imidazol-3-ium bromide (2.87 g, 60 % yield) as a white solid m.p : 182.5 °C.
1H NMR (400 MHz, CDCl3) δ (ppm) 0.85 (t, 3J=7.9 Hz, 2H), 1.21 (s, 12H), 2.06 (qt, 3J=7.6 Hz, 2H), 4.56 (t, 3J=7.3 Hz, 2H), 7.47-7.56 (m, 4H), 7.73 (s, 1H), 7.78 (d, 3J=7.9 Hz, 2H), 11.03 (s, 1H) C18H26BBrN2O2 (393,1262). White solid. Yield = 60 %.
Preparation of 1-(pyrimidin-2-yl)-3-(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)propyl)-1H-imidazol-3-ium bromide (5)
To a mixture of 2-(1H-imidazol-1-yl)pyrimidine (1.02 g, 6.9 mmol, 1.0 eq) and 2-(3-bromopropyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (1.74 g, 6.9 mmol, 1.0 eq) under argon was added acetonitrille (5 mL). The resulting solution was stirred at 100 °C under pressure for 24 hours, after which the product crashed out. The solid material was filtered and washed with dry pentan (5 mL), then collected and dried under vacuo to give the desired product (1.82 g, 4.6 mmol, 66 % yield) as a white solid.
Mp. 236.4 °C
1H NMR (400 MHz, CDCl3) δ (ppm) 0.86 (t, 2H, 3J=7.9 Hz, H9), 1.19 (s, 12H, H11), 2.08 (qt, 2H, 3J=7.6 Hz, H8), 4.76 (t, 2H, 3J=7.2 Hz, H7), 7.59 (m, 1H, H1), 7.89 (d, 1H, 3J=1.5 Hz, H4), 8.20 (d, 1H, 3J=1.8 Hz, H5), 8.88 (d, 2H, 3J=4.8 Hz, H2), 10.72 (s, 1H, H6).
13C NMR (100 MHz, CDCl3) δ (ppm) 7.69 (C9 (HMQC)), 24.4 (C11), 24.6 (C8), 52.0 (C7), 82.9 (C10), 118.3 (C5), 122.2 (C1), 124.1 (C4), 134.9 (C6), 151.6 (C3), 159.5 (C2).
11B NMR (128 MHz, CDCl3) δ (ppm) 33.3 (B). HRMS (ES, [M-Br]+) calcd for [C16H24BN4O2 ]+: 315.1986; found: 315.1986 ESI MS (MeOH) m/z [M-Br]+ calcd for [C16H24BN4O2]+: 315.2; found: 315.0.
CONCLUSION
In conclusion, we have developed a new concept for the design and synthesis of highly active precursor imidazolium salts for the synthesis of N-heterocyclic Carbines used as ligands for the preparation of heterosteroids compounds.
REFERENCES
- O¨ fele, K. J. Organomet. Chem. 1968, 12, P42.
- Wanzlick, H. W.; Sch€onherr, H. J. Angew. Chem., Int. Ed. Engl. 1968, 7, 141.
- Arduengo, A. J.; Harlow, R. L.; Kline, M. J. Am. Chem. Soc. 1991, 113, 361. (b) Arduengo, A. J.; Dias, H. V. R.; Harlow, R. L.; Kline, M. J. Am. Chem. Soc. 1992, 114, 5530.
- Herrmann, W. A. Angew. Chem., Int. Ed. 2002, 41, 1290. (b) Jafarpour, L.; Nolan, S. P. Adv. Organomet. Chem. 2001, 46, 181. (c) Bourrissou, D.; Guerret, O.; Gabbai, F. P.; Bertrand, G. Chem. Rev. 2000, 100, 39. (d) Herrmann, W. A.; K€ocher, C. Angew. Chem., Int. Ed. Engl. 1997, 36, 2162. (e) Nair, V.; Bindu, S.; Sreekumar, V. Angew. Chem. 2005, 117, 1941. (f) Scholten, M. D.; Hedrick, J. L.; Waymouth, R. M. Macromolecules 2008, 41, 7399. (g) McGuinness, D. S.; Suttil, J. A.; Gardiner, M. G.; Davies, N.W. Organometallics 2008, 27, 4238. (h) Raynaud, J.; Ciolino, A.; Baceiredo, A.; Destarac, M.; Bonnette, F.; Kato, T.; Gnanou,Y.; Taton, D. Angew. Chem., Int. Ed. 2008, 47, 5390. (13) Herrmann, W. A.; Elison, M.; Fischer, J.; Koecher, C.; Artus, G. R. J. Angew. Chem., Int. Ed. 1995, 34, 2371–2374.
- Nonnenmacher, M.; Kunz, D.; Rominger, F.; Oeser, T. J. Organomet. Chem. 2007, 692, 2554–2563.
- Taige, M. A.; Zeller, A.; Ahrens, S.; Goutal, S.; Herdtweck, E.; Strassner, T. J. Organomet. Chem. 2007, 692, 1519–1529.
- Ackermann, L.; Althammer, A. Synlett 2006, 3125–3129.
- Huynh, H. V.; Neo, T. C.; Tan, G. K. Organometallics 2006, 25, 1298–1302.
- Scherg, T.; Schneider, S. K.; Frey, G. D.; Schwarz, J.; Herdtweck, E.; Herrmann, W. A. Synlett 2006, 2894–2907.
- Kremzow, D.; Seidel, G.; Lehmann, C. W.; Fuerstner, A. Chem. Eur. J. 2005, 11, 1833–1853.
- Magill, A. M.; McGuinness, D. S.; Cavell, K. J.; Britovsek, G. J. P.; Gibson, V. C.; White, A. J. P.; Williams, D. J.; White, A. H.; Skelton, B. W. J. Organomet. Chem. 2001, 617-618, 546–560.
- Weskamp, T.; Schattenmann, W. C.; Spiegler, M.; Herrmann, W. A. Angew. Chem., Int. Ed. 1998, 37, 2490–2493.
- Weskamp, T.; Kohl, F. J.; Hieringer, W.; Gleich, D.; Herrmann, W. A. Angew. Chem., Int. Ed. 1999, 38, 2416–2419.
- Scholl, M.; Ding, S.; Lee, C. W.; Grubbs, R. H. Org. Lett. 1999, 1, 953–956.
- Sanford, M. S.; Love, J. A.; Grubbs, R. H. J. Am. Chem. Soc. 2001, 123, 6543–6554.
- Herrmann, W. A.; Kulpe, J. A.; Konkol, W.; Bahrmann, H. J. Organomet. Chem. 1990, 389, 85–101.
- Herrmann, W. A.; Kohlpaintner, C. W. Angew. Chem, Int. Ed. 1993, 32, 1524–1544.
- Gardiner, M. G.; Herrmann, W. A.; Reisinger, C.-P.; Schwarz, J.; Spiegler, M. J. Organomet. Chem. 1999, 572, 239–247.
- Schwarz, J.; Herdtweck, E.; Herrmann, W. A.; Gardiner, M. G. Organometallics 2000, 19, 3154–3160.
- L.H. Gade, S. Bellemin-Laponnaz, in: F. Glorius (Ed.), Topics in Organometallic Chemistry, vol. 21, Springer, Berlin, 2007, pp. 117–157; (b) L.H. Gade, S. Bellemin-Laponnaz, Coord. Chem. Rev. 251 (2007) 718–725; (c) M.C. Perry, K. Burgess, Tetrahedron: Asymmetry 14 (2003) 951–961.
- F.E. Hahn, M.C. Jahnke, Angew. Chem., Int. Ed. 47 (2008) 3122–3172; (b) M.S. Sigman, A.D. Jensen, Acc. Chem. Res. 39 (2006) 221–229; (c) W.A. Herrmann, Angew. Chem., Int. Ed. 41 (2002) 1290–1309; (d) D. Bourissou, O. Guerret, F. Gabba, G. Bertrand, Chem. Rev. 100 (2000) 39–92.
- S.T. Liddle, I.S. Edworthy, P.L. Arnold, Chem. Soc. Rev. 36 (2007) 1732–1744; D. Pugh, A.A. Danopoulos, Coord. Chem. Rev. 251(2007) 610–641.
- X.H. Cui, Y.B. Fan, M.B. Hall, K. Burgess, Chem. Eur. J. 11 (2005) 6859–6868; M.C. Perry, X.H. Cui, M.T. Powell, D.R. Hou, J.H. Reibenspies, K. Burgess, J. Am. Chem. Soc. 125 (2003) 113–123; (c) S. Nanchen, A. Pfaltz, Chem. Eur. J. 12 (2006) 4550–4558.
- L.H. Gade, V. César, S. Bellemin-Laponnaz, Angew. Chem., Int. Ed. 43 (2004) 1014–1017.
- R.E. Douthwaite, Coord. Chem. Rev. 251 (2007) 702–717; (b) R. Hodgson, R.E. Douthwaite, J. Organomet. Chem. 690 (2005) 5822–5831.
- T.L. May, M.K. Brown, A.H. Hoveyda, Angew. Chem., Int. Ed. 47 (2008) 7358–7362; (b) M.K. Brown, T.L. May, C.A. Baxter, A.H. Hoveyda, Angew. Chem., Int. Ed. 46 (2007) 1097–1100. and references cited therein.
- P.L. Arnold, M. Rodden, K.M. Davis, A.C. Scarisbrick, A.J. Blake, C. Wilson, Chem. Commun. (2004) 1612–1613.
- H. Hénon, M. Mauduit, A. Alexakis, Angew. Chem., Int. Ed. 47 (2008) 9122– 9124; (b) D. Martin, S. Kehrli, M. d’Augustin, H. Clavier, M. Mauduit, A. Alexakis, J. Am. Chem. Soc. 128 (2006) 8416–8417; (c) H. Clavier, L. Coutable, L. Toupet, J.C. Guillemin, M. Mauduit, J. Organomet. Chem. 690 (2005) 5237–5254.
- Strassner, T.; Ahrens, S.; Zeller, A. WO 2006058535, A2, 2006.
- Lee, K.-M.; Chen, J. C. C.; Huang, C.-J.; Lin, I. J. B. Cryst. Eng. Comm 2007, 9, 278–281.
- Lee, K. M.; Chen, J. C. C.; Lin, I. J. B. J. Organomet. Chem. 2001, 617-618, 364–375.
- Ye, J.; Chen, W.; Wang, D. Dalton Trans. 2008, 4015–4022.
- Chen, C.; Qiu, H.; Chen, W.; Wang, D. J. Organomet. Chem. 2008, 693, 3273–3280.
- Chen, W.; Ye, J. CN 101219399, A, 2008.
- Tanase, A. D.; Herdtweck, E.; Herrmann, W. A.; Kuehn, F. E. Heterocycles 2007, 73, 651–659.
- Wang, C.-Y.; Liu, Y.-H.; Peng, S.-M.; Chen, J.-T.; Liu, S.-T. J. Organomet. Chem. 2007, 692, 3976–3983.
- Tulloch, A. A. D.; Danopoulos, A. A.; Cafferkey, S. M.; Kleinhenz, S.; Hursthouse, M. B.; Tooze, R. P. Chem. Commun. 2000, 1247–1248.
- McGuinness, D. S.; Cavell, K. J. Organometallics 2000, 19, 741– 748.
- Loch, J. A.; Albrecht, M.; Peris, E.; Mata, J.; Faller, J. W.; Crabtree, R. H. Organometallics 2002, 21, 700–706.
- Gruendemann, S.; Albrecht, M.; Kovacevic, A.; Faller, J. W.; Crabtree, R. H. J. Chem. Soc., Dalton Trans. 2002, 2163–2167.
- Tulloch, A. A. D.; Winston, S.; Danopoulos, A. A.; Eastham, G.; Hursthouse, M. B. Dalton Trans. 2003, 699–708.
- Frey, G. D.; Schuetz, J.; Herdtweck, E.; Herrmann, W. A. Organometallics 2005, 24, 4416–4426.
- Yagyu, T.; Oya, S.; Maeda, M.; Jitsukawa, K. Chem. Lett. 2006, 35, 154–155.
- Danopoulos, A. A.; Tsoureas, N.; Macgregor, S. A.; Smith, C. Organometallics 2007, 26, 253–263.
- Catalano, V. J.; Etogo, A. O. Inorg. Chem. 2007, 46, 5608–5615.
- Fiddy, S. G.; Evans, J.; Neisius, T.; Newton, M. A.; Tsoureas, N.; Tulloch, A. A. D.; Danopoulos, A. A. Chem.-Eur. J. 2007, 13, 3652– 3659.
- Li, F.; Bai, S.; Hor, T. S. A. Organometallics 2008, 27, 672–677.
- Scheele, U. J.; Dechert, S.; Meyer, F. Chem.-Eur. J. 2008, 14, 5112–5115.
- Wang, R.; Twamley, B.; Shreeve, J. n. M. J. Org. Chem. 2006, 71, 426–429.
- Wang, R.; Zeng, Z.; Twamley, B.; Piekarski, M. M.; Shreeve, J. n. M. Eur. J. Org. Chem. 2007, 655–661.
- Wang, H. M. J.; Lin, I. J. B. Organometallics 1998, 17, 972.
- Baskakov, D.; Herrmann, W. A.; Herdtweck, E.; Hoffmann, S. Organometallics 2007, 26, 626.
- G. Occhipinti. ; H. R. Bjørsvik. ; K. W. Tornroos. ; A. Furstner. ; and V. R. Jensen. Organometallics, 2007, 26, 4383.
- Xie, Y.;Pi, S.; Wang, J.; Yin, D.; Li, J. J. Org. Chem. 2006, 71, 8324.
- Molander, G.A.; Yun, C-S.; Ribagorda, M.; Biolatto, B. J. Org. Chem. 2003, 68, 5534.







ISSN Online: 2231-5039

![]()




{kind=link}