Calculation of Molecular Lipophilicity and Drug Likeness for Few Heterocycles
Download this article as:
![]()
Log P value for 16 vinyl, nitrogen and sulphur heterocycles were calculated using eight computer programs: HyperChem 7.0 (based on atom contributions), XLOGP (based on atom contributions),KowWin (based on atom/fragment contributions),CLOGP (based on fragmental contributions), ALOGPS 2.1 (based on atom-type electro topological-state indices and neural network modeling), and IA logP (based on atom-type electrotopological-state indices and neural network modeling), miLogP using Molinspiration and MolSoft. The drug likeness has been calculated using Molinspiration and MolSoft programs. Almost all the heterocycles were found to obey Lipinski’s rule of 5 for an orally active drug.
KEYWORDS:Molecular lipophilicity; drug likeness and heterocycles
Introduction
The partition coefficient for octanol–water (log Pow) has become the preferred measure for lipophilicity in the development of biologically active molecules, in which transport across biological membranes is often critical . The widespread application of lipophilicity to drug design explains the need for quick procedures to quantify molecular lipophilicity, particularly at the screening level 4-5. Giving consideration to the significance of log P, this paper is focused on the theoretical determination of log P and other molecular properties like refractivity, polarisability, polar surface area, log S, molecular volume, drug likeness and number of rotatable bonds ,using computer programs.
Material and Methods
Log P value for 16 vinyl nitrogen and sulphur heterocycles (Fig 1)were calculated using eight computer programs: Hyper Chem 7.0 (based on atom contributions)6, XLOGP (based on atom contributions7), KowWin (based on atom/fragment contributions8, CLOGP (based on fragmental contributions 9, ALOGPS 2.1 (based on atom-type electro topological-state indices and neural network modeling)10, IA logP (based on atom-type electrotopological-state indices and neural network modeling), miLogP using Molinspiration and MolSoft. The drug-likeness has been calculated using Molinspiration and MolSoft programs.
Calculation of Lipophilicity (log P)
In our work, eight computer programs based on different calculation methods for computing log P have been compared.
Hyperchem 7.0.
The computer program Hyper Chem 7.0 calculates log P values according to Ghose, Prichett and Crippen11.Their method avoids correction factors and estimates lipophilicity on the individual atom basis. The program lists 51 atom contributions for each atom-type. The log P value is estimated by summing up all atom contributions.
XLOGP
The XLOGP is another atom-additive method, based on summation of atomic contributions, but it includes ten additional correction factors for some intramolecular interactions. Atoms are classified by their hybridization states and neighboring atoms.
(Logkow) kowwin
The LogKow (KowWin) program calculates log P values of organic chemicals using the atom/ fragment method. The log P value of the calculated compounds is by simply summing up all atom/ fragment contribution values and correction factors. CLOGP The CLOGP program is based on the fragmental method developed by Leo and Hansch.
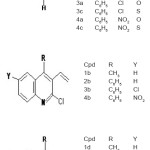 |
Figure 1 Click here to View figure |
ALOGPS 2.1
The ALOGPS 2.1 package included a program for predicting the lipophilicity of chemical compounds. A method for predicting log P values is based on atom-type electro topological state (E-state) indices and neural network modeling developed by I. V. Tetko et al.,10
IA logP
The IA logP program predicts lipophilicity of compounds using neural network algorithms and E-state atom indices. The IAlogP predictor was developed by Interactive Analysis, using neural networks technology to derive a set of 10 fold cross-validated networks.
Molinspiration
LogP (octanol/water partition coefficient)
LogP is calculated by the methodology developed by Molinspiration as a sum of fragment-based contributions and correction factors. Method is very robust and is able to process practically all organic and most organometallic molecules.
Milog P
Method for logP prediction developed at Molinspiration is based on group contributions. These have been obtained by fitting calculated logP with experimental logP for a training set more than twelve thousand, mostly drug-like molecules.
Molecular property predictions using MolSoft
In MolSoft all molecular property predictors are calculated using fragment-based contributions. Other properties calculated
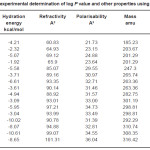 |
Table 1: Results of experimental determination of log P value and other properties using Hyperchem 7.0 Click here to View table |
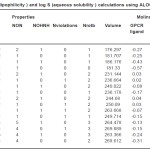 |
Table 2: Log p (lipophilicity ) and log S (aqueous solubility ) calculations using ALOGPS 2.1 program Click here to View table |
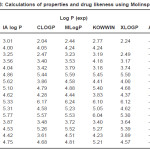 |
Table 3: Calculations of properties and drug likeness using Molinspiration Click here to View table |
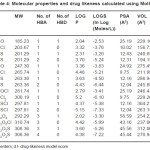 |
Table 4: Molecular properties and drug likeness calculated using MolSoft Click here to View table |
HYDRATION ENERGY kcal/mol
Hydration energy is very important in the selectivity filter of ion channels where the drug is almost entirely stripped from the hydration water.
Refractivity A3
There are several potential classes of parameters used in QSAR studies. The selection of parameters is an important first step in any QSAR study. If the association between the parameter(s) selected and activity is strong, then activity predictions will be possible. One such important parameter is refractivity.
Polarisability A3
A challenge facing the practical application of computational alchemy to drug discovery is insuring the adequacy of the underlying potential energy function. Although existing potential functions that assume pairwise additivity of nonbonded interatomic interactions appear to be sufficient in many cases the description of certain interactions may require explicit treatment of electronic polarisability.
Molecular Polar Surface Area TPSA is calculated based as a sum of fragment contributions. O- and N- centered polar fragments are considered. Briefly, the procedure is based on the summation of tabulated surface contributions of polar fragments.
nviolations -”Rule of 5″ Properties is set of simple molecular descriptors used by Lipinski in formulating his “Rule of 5”. Molecules violating more than one of these rules may have problems with bioavailability.
nrotb-Number of Rotatable Bonds
This simple topological parameter is a measure of molecular flexibility. It has been shown to be a very good descriptor of oral bioavailability of drugs.
Molecular Polar Surface Area PSA
Molecular polar surface area (PSA) is a very useful parameter for prediction of drug transport properties.
Molecular volume
Molecular volume determines transport characteristics of molecules, such as intestinal absorption or blood-brain barrier penetration. Volume is therefore often used in QSAR studies to model molecular properties and biological activity.
Molecular properties predictions using MolSoft
Drug-likeness score predicts an overall drug-likeness score using a Support Vector Machine (SVM) classifier and MolSoft’s fingerprints. Drug likeness may be defined as a complex balance of various molecular properties and structure features, which determine whether particular molecule is drug or non-drug.
Discussion
In the relation between the structure and activity, lipophilic character log P appears to be among the most important physicochemical parameter. This paper is intended to determine the lipophilicity of 16 vinyl nitrogen and sulphur heterocycles.
It is evident from the results of HyperChem calculation of log P (Table I) that the nitro substituted compounds have a negative log P as is expected from the nature of the nitro group. The compounds 3b, 3c and 2b are found to have high log P values compared to the other compounds. It is also seen from the calculations that the refractivity of the phenyl substituents is greater than the 1series of compounds. So is the polarisibility of the compounds. The log P (exp) calculated by all other programs (Table II) also show the same trend – the value being highest for 3b compound. Compounds 2a and 2b show a positive value for kinase inhibitor score (Table III). Greater the score, greater is the inhibiting activity. Similarly compounds 2a, 2b, 3a and 3b show a positive value for GPCR ligand score.
2a,3c,2c compounds showed a positive value and hence more drug likeness as seen from the calculations of MolSoft program(Table IV). In order to increase the drug likeness score various modifications were tried with the compound 2c and the score was found to increase from 0.5 to 0.84 when the structure 2c had chloro substituents at 5 and 7 positions in the quinoline ring. All the computer programs were found to be relatively simple and applicable to QSAR studies. The programs are highly useful for predicting the solvation properties.
Acknowledgements
The authors sincerely thank the authorities of Avinashilingam University for Women, Coimbatore for providing facilities to carry out this work.
References
- Carrupt P. Testa B. and Gaillard P., in Reviews in Computational Chemistry, K. B. Lipkowitz and D. B. Boyd, Eds., Wiley–VCH, New York, 1899, 11, 241(1997).
- Hansch C. and Leo A., Exploring QSAR Fundamentals and Applications in Chemistry and Biology; American Chemical Society, Washington, D.D. (1995).
- Martin Y. C., Quantitative Drug Design: A Critical Introduction, Marcel Dekker, New York (1978.)
- Martin Y. C., Quantitative Drug Design: A Critical Introduction, Marcel Dekker, New York (1978.)
- Mannhold R. and Dross K., Quant. Struct.- Act. Relat., 15: 403 (1996).
- van de Waterbeemd H. and Mannhold R., Quant. Struct.-Act. Relat. , 15: 410 (1996).
- Ghose K., Pritchett A., and Crippen G. M., J. Comput.Chem. 9: 80 (1988).
- Wang R., Fu Y., and Lai L., J. Chem. Inf. Comput. Sci. 37: 615 (1997).
- Meylan W. M. and Howard P. H., J. Pharm. Sci. 84 (1995).
- Chou J. T. and Jurs P. C., J. Chem. Inf. Comput. Sci. 19: 172 (1979).
- Tetko V., J. Chem. Inf. Comput. Sci., 42 , 717 (2002).







ISSN Online: 2231-5039

![]()




{kind=link}